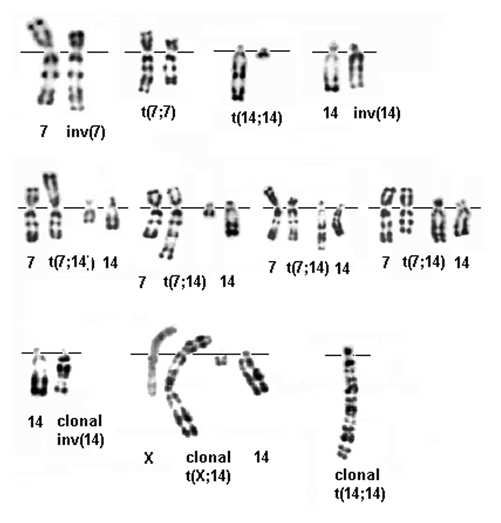
Imagen 4 con fines Didácticos: Reordenamientos esporádicos (filas 1 y 2) y clonal (fila 3) en la AT (R-banding). Fila 1, de izquierda a derecha: inv (7) (p14q35), t (7, 7) (p14; q35), t (14; 14) (q11; q32), inv (14) (q11q32); Row 2 :, de izquierda a derecha: t (7; 14) (p14; q11), t (7; 14) (q35; q11), t (7; 14) (p14; q32), t (7; 14) ( q35; q32); Row 3, de izquierda a derecha: inv (14) (q11; q32), t (X; 14) (q28; q11) (tenga en cuenta a finales de los X replicar a la izquierda), t (14; 14 ) (q11; q32) – Tomado de Médecine / Ciencias 1986; 2: 298-303.
Los individuos con AT pueden también tener una mayor frecuencia de rompimientos espontáneos en sus cromosomas, así como una mayor frecuencia de reacomodos de cromosomas. Estas anormalidades ocurren frecuentemente en la cercanía de genes esenciales para la función de linfocitos, tales como los genes receptores de antígenos de inmunoglobulina y linfocitos T. La frecuencia de rompimientos de cromosomas (véase la imagen adjunta) aumenta cuando los linfocitos T son expuestos a rayos-X en el laboratorio, y esto conforma las bases para una prueba de diagnóstico especializada para la AT.
Schubert et al estudiaron las deficiencias en los subtipos de células T CD4+ y CD8+ en la AT. Encontraron que los linfocitos CD45RA+ (“naive” o vírgenes – emigrantes tímicos tempranos) se encuentran fuertemente disminuidos, mientras que los NK (CD56+) están significativamente elevados en estos enfermos. Utilizando varios rangos de referencia pediátricos quedan únicamente como claramente anómalos en esta paciente los bajos niveles de la población CD45RA+.
El análisis citogenético mostrará en los homocigotos y portadores alteraciones cromosómicas espontáneas con una elevada frecuencia. Dichas alteraciones serán de utilidad como un elemento confirmatorio más, o diagnósticas, en aquellos raros casos (menos del 5%), donde los niveles de alfa fetoproteína no se encuentran elevados.
La identificación de fragmentación del ADN postirradiación ha permitido el diseño de técnicas de cribado para los heterocigotos de relativa sencillez. Por otra parte, la detección precisa de la alteración génica representa un gran desafío, derivado del alto número de modificaciones (mayores a 400 descritas) distribuidas en los 66 exones del gen ATM. Son, por tanto, atribución de escasos laboratorios en el mundo.
El interés creciente en detectar el estado de portadores (heterocigotos) va más allá del posible consejo genético en una enfermedad de muy baja frecuencia, para relacionarse con el riesgo aumentado a neoplasia en los portadores.
La AT pertenece a un grupo de enfermedades colectivamente denominadas “síndromes de inestabilidad genómica” dentro de las cuales y aparte de la AT, encontramos: el síndrome de Nijmegen, el síndrome de Seckel, la anemia de Fanconi, el xeroderma pigmentoso, el síndrome de Bloom y otras 5 infrecuentes enfermedades. Siendo los mecanismos diferentes en cada caso, todas ellas conllevan la incapacidad de detectar la lesión del ADN, transmitir el mensaje o finalmente, reparar el daño sufrido en el mismo. El presente caso puede servir de alerta y motivo de atención para estas enfermedades.
•Tratamiento general: Aún no existe cura para ninguno de los problemas de la AT, y el tratamiento es de apoyo en gran medida. Se debe fomentar la participación de los pacientes en tantas actividades como sea posible. Se debe animar a los niños a ir a la escuela de forma regular y deben recibir apoyo para mantener un estilo de vida tan normal como sea posible. Se deben incluir terapeutas físicos y ocupacionales en el equipo de tratamiento para prevenir el desarrollo de rigidez en los músculos y para mantener movilidad funcional. Se debe buscar un diagnóstico temprano e instituir terapia específica en cualquier sospecha de infecciones.
Para los pacientes que tienen niveles normales de inmunoglobulinas en suero y respuestas normales de anticuerpos a vacunas, las vacunas con influenzae y neumococos pueden ser de gran ayuda. Para pacientes con deficiencias totales de IgG y deficiencias subclase IgG, y/o los pacientes que tienen problemas produciendo respuestas normales de anticuerpos a vacunas, puede indicarse terapia con inmunoglobulina.
Se debe prestar atención específica a los problemas de deglución. Los pacientes que aspiran o tienen alimentos y líquidos entrando en la tráquea y pulmones pueden mejorar cuando se eliminan los líquidos no espesos de la dieta. En algunos individuos, puede ser necesario un tubo desde el estómago al exterior del abdomen (fístula gástrica), para eliminar la necesidad de deglutir amplio volúmenes de líquidos y para disminuir el riesgo de aspiración.
Deben limitarse las radiografías en diagnósticos dado el riesgo teórico de que las radiografías pueden provocar un rompimiento de cromosomas que resulte en el desarrollo de neoplasias. En general, las radiografías sólo deben realizarse si el resultado influenciará la terapia y no existe otra manera de obtener la información que se obtendrá con radiografías.
•Terapia específica: En este momento no es posible la terapia específica para la AT. El uso de trasplantes de timo, hormonas de timo y trasplante de médula ósea no ha llevado a ninguna mejora. De la misma manera, no hay evidencia de que alguna terapia de nutrición específica sea de beneficio. Sin embargo, ahora que se ha identificado el gen y se está estudiando la función normal del gen, se espera que pronto se encuentre disponible una terapia nueva y específica.
Bibliografía
- Syllaba L, Henner K. Contribution a l’independence de l’athetose double idiopathique et congenitale. Rev Neurol 1926; 1: 541-62.
- Louis-Bar D. Sur un syndrome progresiff comprenant des télangiectasies capillaries cutanées et conjontivales symétriques à disposition naevoîde et des troubles cérébelleux. Confin Neurol. 1941; 4: 33-42.
- Boder E, Sedgwick RO. Ataxia-telangiectasia: a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia, and frequent pulmonary infection. Univ S Calif Med Bull 1959; 9: 15-27.
- Shiloh Y (1997) Ataxia-telangiectasia y el síndrome de ruptura de Nijmegen: trastornos relacionados, pero aparte de los genes. Annu Rev Genet 31 : 635 – 662.
- Rebollo MA. Ataxia-telangiectasia. Acta Neurol Latinoamer 1961; 7: 331-336.
- Expert Reviews in Molecular Medicine 2003. Cambridge University Press.
- Sadighi Akha AA, et al. Gammapatía oligo / monoclonal e hipergammaglobulinemia en ataxia-telangiectasia. Un estudio de 90 pacientes. Medicina (Baltimore) 1999; 78 (6): 370–81. doi: 10.1097 / 00005792-199911000-00002.
- Schubert R, Reichenbach J, Zielen S. Deficiencies in CD4+ and CD8 T cell subsets in ataxia telangiectasia. Clin Exp Immunol 2002; 129 (1): 125-32.
- Marcos J, García-Güelfi A, Maggi R, Sande MT. Ataxiatelangiectasia (enfermedad de Louis-Bar). Arch Ped Urug 1962; 33: 530-6.Gatti RA. Ataxia-telangiectasia. In: Scriver CR, Beaudat AL, Sly WS. Metabolic and Molecular Basis of Inherited Diseases. 8ª ed. New York: McGraw-Hill, 2001: 705-32.
- Dres. Marcos Delfino, Rina Bruzzone, Andrea Rey, Aurora Delfino, Ma. Catalina Pírez. Ataxia-telangiectasia (síndrome de Louis-Bar). Arch Pediatr Urug 2006; 77(2): 154-158.
- Gatti RA, Berkel I, Border E. Localisation of the ataxiatelangiectasia gene to chromosome 11q22-23. Nature 1988; 336: 577-80.
- Swift A, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991; 325: 1831-6.
- Boder E, Sedgwick RO. Ataxia-telangiectasia: a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia, and frequent pulmonary infection. Univ S Calif Med Bull 1959; 9: 15-27.