En 1997 el grupo de Shiloh identificó en esa región el gen mutado de la AT, es el gen ATM. El producto de ese gen resultó ser una proteinquinasa con funciones de “señal” que interviene en el control del ciclo celular, la recombinación de ADN, la apoptosis y otras respuestas celulares al daño de ADN.
Las anormalidades en este control mediado por ATM producen ADN dañado, con lo cual se acumulan las roturas cromosómicas con el paso del tiempo y la célula muere. Estos fenómenos afectarían especialmente los timocitos, los linfocitos B inmaduros, las células de Purkinje del sistema nervioso central y el endotelio vascular.
La marcha atáxica puede estar presente en el segundo año de vida; a los cuatro años de vida la presentará el 85% de los afectados. La ataxia afecta progresivamente la musculatura axial, de las extremidades, del paladar surgiendo así disartria, babeo, apraxia ocular, coreoatetosis. A los 10 años de vida la marcha independiente es imposible.
Las telangiectasias surgen posteriormente, entre los 2 y los 8 años de edad en escleróticas y piel (orejas, mejillas, pliegues de codos, cuello, etcétera), se vuelven aparentes después del comienzo de la ataxia. La hipersensibilidad cutánea a la luz se evidencia por un envejecimiento temprano de la piel. La inmunodeficiencia celular y humoral es responsable de la recurrencia de las infecciones respiratorias altas y bajas, estas últimas conducen a enfermedad pulmonar crónica. Las neumonías infecciosas (virales o bacterianas) o por broncoaspiración constituyen la primera causa de muerte en estos enfermos.
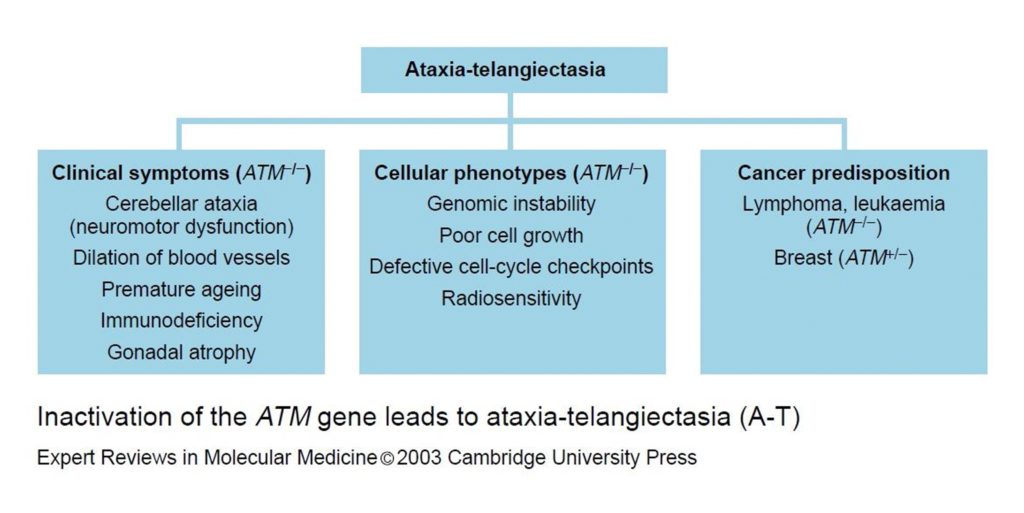
Figura 2.- Inactivación del gen de ATM que lleva a la ataxia-telangiectasia. La figura resume el los síntomas clínicos, fenotipos celulares y predisposición de cáncer de individuos con las mutaciones en el gen ATM.
La segunda causa de muerte son los cánceres. El 38% de los pacientes con esta enfermedad desarrollan un neoplasma en su vida. Esto puede ser debido a las translocaciones cromosómicas y roturas excesivas del ADN que caracterizan la inestabilidad genética de esta enfermedad. Un 85% de estos neoplasmas son de origen hematológico. La mayoría son tumores linforreticulares, desarrollando uno de cada 100 afectos un nuevo tumor anual a partir de los 10 años. Generalmente se trata de linfomas (60%) y leucemias agudas (27%), aunque después de los 20 años la frecuencia de estos cánceres cae un 50%, siendo más frecuentes los tumores de origen epitelial. La hipersensibilidad a la radiación ionizante debida a la inestabilidad genética hace que los estudios imagenológicos en estos pacientes deban ser restringidos y que las dosis de radiación para tratar estos tumores sean menores a las usuales.
El riesgo de cáncer (especialmente de mama) entre portadores no está aún confirmado. Estudios realizados por Swift et al sugieren que hasta un 5% de todos los pacientes que mueren de cáncer menores de 45 años podrían ser portadores del gen ATM (heterocigotos). Las madres de los pacientes con síndrome de Louis-Bar, portadoras del gen ATM, tendrían un riesgo incrementado para cáncer de mama.
El timo puede ser indetectable desde la primera infancia y si está presente es pequeño y carece de diferenciación corticomedular; las amígdalas son hipoplásicas, así como el tejido linfoide periférico. En la sangre hay linfopenia progresiva tanto para los linfocitos T como B; los
linfocitosTCD4+ (timodependientes) mueren selectivamente, lo que finalmente conduce a una inversión de la normal relación CD4-CD8. Las funciones de uno y otro disminuyen gradualmente, lo que sería también un efecto de la excesiva rotura de ADN. Los defectos de tipo humoral incluyen deficiencia de inmunoglobulinas.
Existe un déficit de IgA tanto a nivel sérico como de la IgA secretoria (por ejemplo, saliva) en el 60% de los casos. La IgG2 disminuye en el 80%; también pueden disminuir la IgE, las isohemaglutininas y las respuestas de anticuerpos específicos. La edad de fallecimiento es generalmente alrededor de los 20 años; casi todos los pacientes mueren antes de los 39 años.
•Manifestaciones neurológicas: AT es similar y sin embargo diferente de otros trastornos relacionados con el cerebelo. Tanto AT y las enfermedades cerebelosas clásicos comparten la característica común de control anormal del movimiento coordinado. De alguna manera, sin embargo, los pacientes con AT parecen haber relativamente preservada funciones del cerebelo, por lo menos hasta la primera década o más.
Al mismo tiempo, hay anomalías prominentes del movimiento de los ojos, postura, la marcha, el movimiento y el habla que son diferentes de los observados con daño cerebelar pura. Algunas de estas características serán revisadas.
Las partes del sistema nervioso que no sufren la AT:
- -Audición
- -El sentido del tiempo, el lugar y el propósito
- -Capacidad para comprender las interacciones sociales
- -Visión (pero no de los movimientos oculares)
Es importante tener en cuenta que AT no afecta a todo el cerebro. Algunas áreas del cerebro que son especialmente vulnerables en AT, pero otras áreas funcionan normalmente durante décadas en la mayoría o en todos los pacientes. Algunos de los sistemas del cerebro que parecen ser particularmente resistente a los daños incluyen las primeras etapas de la visión en la retina y el cerebro; audiencia en todos los niveles de procesamiento; equilibrio (Esto es diferente de la postura como se describe a continuación); las etapas finales de los movimientos oculares (descrito más adelante), y en muchos pacientes, las funciones corticales que permiten la comprensión de las interacciones sociales y la orientación en tiempo, lugar y propósito. Este daño cerebral selectivo es sorprendente, ya que la proteína ATM se produce en todas las células del cerebro. La comprensión de por qué la pérdida de ATM es esencial para la función normal en algunas zonas, pero no todas las áreas pueden ayudarnos a entender cómo la pérdida de ATM daña el cerebro. Esa comprensión puede a su vez conducir al desarrollo de tratamientos que reduzcan al mínimo los daños.
Otra característica importante de AT es que los pacientes individuales difieren en la gravedad de los diferentes trastornos neurológicos. Si bien la naturaleza de cada deficiencia es a menudo muy distintivo, los niños difieren entre sí en la cantidad de cada deficiencia se presenta a una edad determinada. La figura 2 ilustra los distintos «forma» del deterioro neurológico en siete pacientes con AT, que son aproximadamente de la misma edad. Por ejemplo, un niño puede tener problemas graves con la alimentación y deglución al tener menos deterioro de la marcha. Otro niño puede tener dificultad notable con control de la cabeza, mientras que el tercero puede tener el mayor deterioro de la marcha. A pesar de esta variabilidad global entre los diferentes niños, cuando los hermanos tienen AT, los perfiles de sus deficiencias neurológicas tienden a ser similares en la «forma».
La postura, la marcha, el tono y el movimiento: Los niños con AT suelen adoptar posturas inusuales con la cabeza o el tronco. La cabeza influir a una posición lateral, hacia delante o hacia atrás extrema, y permanecer en esta posición durante un máximo de medio minuto antes de corregir, a menudo en una posición descentrada diferente. La misma anormalidad postural se puede ver con el tronco cuando en una posición sentada. Aquí el límite está en el margen de estabilidad, tiene los brazos y las piernas se extienden en el lado opuesto para evitar el vuelco. Un padre describió: «Porque un niño con ataxia, que tiene el mejor balance» Algunos niños con AT nunca desarrollan esta postura, sin embargo, cuando está presente, se produce sólo en los niños más pequeños. Por el contrario, los niños mayores y adultos jóvenes han aumentado el tono o rigidez del tronco y el cuello. No está claro si este problema postural inusual desaparece porque mejora, o porque se sustituye por el aumento en el tono troncal.
El andar en AT es a menudo distintivo. Los niños normalmente a pie con sus pies inapropiadamente cercano uno del otro y que tienden a cruzar sus pies. A menudo prefieren caminar rápido o correr, y en ocasiones pueden caminar mientras se inclina hacia adelante sobre los dedos de los pies. Es muy curioso que la mayor inestabilidad al estar de pie o caminar muy lentamente.