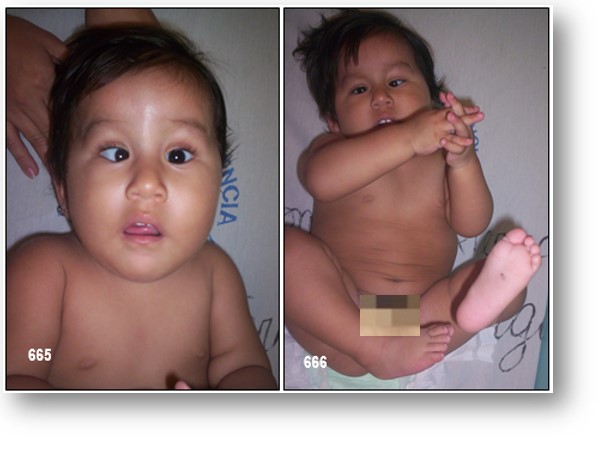
INFORMACIÓN BÁSICA.- Síndrome de Moebius.- (OMIM 157900).- El síndrome de Moebius (SM) es una enfermedad congénita caracterizada por parálisis facial congénita no progresiva, simétrica (Diplejía facial) o asimétrica, con ausencia de abducción de los ojos debida a lesión del VI y VII nervios craneales, en asociación con otras alteraciones neurológicas, músculo-esqueléticas, craneofaciales, respiratorias, y ortopédicas, entre otras. Se trata de un trastorno congénito raro, por lo cual no es fácil encontrar datos estadísticos respecto a su incidencia, que se calcula en torno a 1 caso por cada 50.000 nacidos vivos. Hasta la fecha, se han reportado aproximadamente 300 casos en la literatura. La parálisis congénita del nervio facial y abducens fueron descritas por Von Graefe en 1880, seguido por Harlam en 1881 y Christholm en 1882. Sin embargo, fue un neurólogo alemán, Paul Julius Moebius, quien notó la asociación de dicha parálisis facial congénita con otras malformaciones y realizó una comparación con casos previamente descritos, elaborando la clásica descripción del síndrome inicialmente conocido como Infantile Kernschwund y que posteriormente llevó su nombre. En los últimos años se han asociado una gran variedad de malofrmaciones con este síndrome, lo que ha hecho de él un síndrome con alta variabilidad, con diferentes presentaciones, sin una etiología o patología bien definida.
La primera manifestación de la enfermedad es la dificultad que tienen los bebés para la succión. Los recién nacidos afectados también pueden presentar babeo excesivo y estrabismo. Más adelante, los signos que dominan el cuadro clínico son: ausencia de expresión facial, incapacidad para sonreír, ausencia de parpadeo, y limitación de los movimientos laterales de los ojos (cara de máscara). Otras anomalías pueden también estar asociadas, como, por ejemplo, deformación de la lengua, que conduce a dificultades para hablar, y anomalías de la mandíbula. Las malformaciones de las extremidades se observan en un tercio de estos pacientes y pueden incluir pie zambo, dedos ausentes o unidos, y anomalía de Poland (hipoplasia del pectoral). La inmensa mayoría de los niños padecen hipotonía muscular, especialmente en la parte superior del cuerpo, lo que produce un retraso en el aprendizaje de la marcha. En aproximadamente un 10 por ciento de los casos se detecta un ligero déficit intelectual.
El SM es debido a una anomalía en el desarrollo del séptimo par craneal (nervio facial) en todos los pacientes, y del sexto par craneal (nervio motor ocular externo) en el 75% de los casos. Ocasionalmente, otros nervios craneales pueden también estar afectados (el III, IV, V, IX, X y/o XII par craneal).
La mayoría de los casos de SM son esporádicos sin ningún antecedente familiar (idiopático), aunque se ha asociado a la ingesta de misosprostol durante el embarazo, y a otras sustancias teratógenas como el alcohol, cocaína, talidomida, misoprostol (Cytotec® 200mcg), etc. Precisamente Calderón León M, describe en nuestro país un caso clínico de una paciente de 11 meses de edad cuya madre usó tabletas de misoprostol por vía vaginal (400 mcg), durante el primer trimestre de gestación, con fines abortivos, presentando al nacimiento defectos congénitos.
El misoprostol es un análogo de la prostaglandina E1 y fue creado para el tratamiento de la úlcera gástrica, pero, además posee acción uterotónica y tiene capacidad de maduración del cuello uterino. Si se lo usa al final de la gestación colabora como buen inductor del parto, no siendo así en el primer trimestre de gestación, donde los efectos son devastadores provocando un aborto, o en su defecto, genera déficit en la irrigación sanguínea, afectando el desarrollo embriológico del nuevo ser. En países donde el aborto no es legal, el misoprostol se emplea para la interrupción del embarazo, sin embargo la infrecuente supervisión profesional, llevan a que algunos de los embarazos continúen produciendo exposición prenatal al misoprostol, lo cual está asociado con la ocurrencia de anomalías congénitas.
Por otro lado, la enfermedad no es progresiva y el tratamiento es esencialmente sintomático. Los niños pueden requerir de biberones especiales o de sondas de alimentación para lograr una nutrición adecuada. Los pacientes pueden también beneficiarse de terapia física y logopedia para mejorar sus habilidades motoras y su coordinación, así como para conseguir un mayor control del lenguaje y de la alimentación.
El estrabismo generalmente se corrige con cirugía y las malformaciones de las extremidades y de la mandíbula se pueden también mejorar generalmente con intervención quirúrgica.
El injerto de músculo puede proporcionar una cierta movilidad facial, incluyendo la capacidad para sonreír.
Clasificación.- Existen 3 etiologías propuestas por 3 autores diferentes para tratar de entender la enfermedad: • Legum y col. propusieron un defecto genético en el desarrollo del romboencéfalo, incluyendo el origen del VII nervio craneal.
(1) • Bavinck y Weaver describieron una interrupción del aporte vascular en el territorio de la arteria subclavia entre la sexta y séptima semanas de gestación, con isquemia del núcleo de nervio facial.
(2) • Por último, la exposición a teratógenos, como ya lo hemos descrito en líneas anteriores, durante el embarazo puede ser causa de las 2 hipótesis previas. El ejemplo más claro es el uso como abortivo no electivo del misoprostol, un análogo prostaglandínico E1, durante el primer trimestre del embarazo. Debido a la heterogenicidad del síndrome se han propuesto diferentes clasificaciones para unificar criterios. Una muy conocida es la publicada por Abramson, Cohen y Mulliken quienes utilizan el acrónimo CLUFT, por sus siglas en inglés, para malformaciones en el nervio craneal (craneal nerve), extremidad inferior (lower limb), extremidad superior (upper limb), cara (face) y tórax (thorax).
(3) •Dentro de las malformaciones craneofaciales se han descrito: telecanto, microgenia o micrognatia, defectos del pabellón auricular y paladar hendido, entre otras. Las malformaciones de tipo músculo-esqueléticas abarcan: sindactilia, mano hendida, pie equino varo y síndrome de Poland como las más frecuentes. De la misma forma, hay malformaciones oftalmológicas com estrabismo y defectos de refracción, y neurológicas como epilepsia y calcificaciones cerebrales. En el año 2002, la Dra. Terzis propuso una nueva clasificación que permite al cirujano identificar deficiencias en los pacientes y establecer una propuesta quirúrgica:
(4) – Grupo A: conocido como SM Clásico, con parálisis facial bilateral completa y parálisis del nervio abducens. – Grupo B: o SM Incompleto, caracterizado por parálisis facial congénita con movimiento residual unilateral. – Grupo C: conocido como SM Like o similar a Moebius, en donde los pacientes tienen parálisis facial unilateral pero otros nervios craneales afectados. En los estudios de genética, el SM frecuentemente se ha reportado como una patología esporádica; sin embargo, algunos casos se han asociado con herencia autosómica dominante, recesiva o ligada al X. Estudios citogenético de bandas GTG sugieren alteraciones en dos loci: 13q12.2-13 y 1p22.
En Ecuador, según nuestra revisión, existen publicaciones esporádicas, sin contar hasta el cierre de esta edición que nuestro país cuente por lo menos con un catastro de este tipo de enfermedades poco frecuentes o raras.
La serie más actual y más extensa en el siglo XXI era la serie italiana publicada por Arturo Carta en el 2011, donde reúne 55 pacientes para realizar una descripción de las manifestaciones oftálmicas, principalmente.
En 2014, Borbolla y col. del Servicio de Oftalmología Pediátrica del Instituto Nacional de Pediatría de México publicaron una serie de 64 casos en la que describen principalmente manifestaciones oftalmológicas, siendo la serie más grande publicada hoy en día. Arrieta JP et al., nuestra una serie, junto con el apoyo de múltiples servicios dentro de un solo centro hospitalario, lograron identificar 115 casos así como las manifestaciones clínicas presentadas en cada uno de ellos. El equipo de John Mulliken y col., en 1998 publican una serie de 27 pacientes en una sola unidad hospitalaria y proponen la nemotecnia CLUFT por las siglas en inglés cranial nerve, lower limb, upper limb, face and thorax malformation para el estudio de estos pacientes, con la intención de lograr una integración para su estudio de forma interinstitucional y homogeneizar los términos. De forma similar a lo reportado por el grupo de Arrieta et al., sus pacientes tenían compromiso del VII y del VI nervios craneales. Tanto el estudio de Mulliken como el de Terzis en el 2002 coinciden en que junto con la parálisis facial, los pares craneales V y XII son los más afectados; seguidos por los demás en porcentajes similares. Estas cifras contrastan con los hallazgos de Arrieta et al,. en donde el nervio hipogloso (XII) y el patético/troclear (IV) son los más afectados en el 13.9% y el 7.8% respectivamente, pero con diferencia en el número de casos entre los 27 de Mulliken, los 43 de Terzis y los 115 de la serie de Arriesta et al., . Tal y como describen la mayoría de las publicaciones en la literatura, como las de Terzis, Carta y Ramos de Souza-Dias, el estrabismo fue la manifestación clínica más frecuente en el 62.6% de los casos y los defectos refractivos aparecieron en el 13% de los casos. Muchos autores coinciden que las manifestaciones craneofaciales más frecuentes son el telecanto, el paladar hendido y el pie equino varo como manifestación musculoesquelética más frecuente en el 46.1% de los casos, seguido de la sindactilia y del síndrome de Poland, entre otros. El SM asociado a craneosinostosis no sindrómica, esferocitosis y amelogénesis imperfecta, no había sido reportado previamente en la literatura. La relación del consumo de misoprostol con el diagnóstico de SM sigue siendo un tema de incertidumbre e investigación. En 1998, Pastuszak y col. en un estudio multicéntrico de 96 pacientes describen que 47 (49%) tuvieron madres que tomaron misoprostol durante el primer trimestre del embarazo.
Es importante el asesoramiento genético. La inmensa mayoría de casos se dan de forma aislada, como caso único en una familia en la que no existe ningún otro miembro con el síndrome. Esto significa que el riesgo de recurrencia es muy bajo (1%).
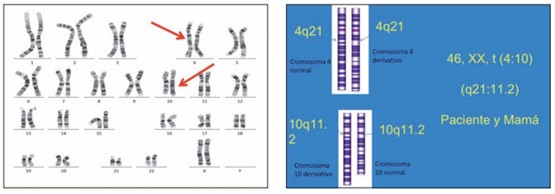
Por otra parte, la mayoría de las publicaciones de SM hacen referencia a que no hay evidencia suficiente para apoyar un patrón de herencia autosómico dominante, sino más bien mutaciones de novo. Ziter y col. en 1977 presentaron una translocación recíproca entre el cromosoma 1p34 y el cromosoma 13q13, así como Slee y col., en 1991 quienes publican una deleción en el cromosoma 13q22.2, lo que suponía que el daño podría estar en el cromosoma 13q22.2 – q13, por lo que Uzumcu y col. en 2009, realizan un estudio molecular para buscar esas mutaciones en 9 pacientes sin encontrarlas. En el estudio de Arriesta et al., encontraron una translocación no antes reportada entre el cromosoma 4 y el 10, desafortunadamente sin tener resultados del estudio molecular.
Existen casos familiares de parálisis facial que se transmite con patrón autosómico dominante.
Estos casos son poco frecuentes, y la manifestación clínica del síndrome es un poco distinta, ya que los afectados presentan únicamente parálisis facial, sin las otras anomalías que suelen acompañar habitualmente al SM. Aunque en algunas publicaciones científicas aún se siguen encuadrando bajo el nombre de SM, actualmente estos casos familiares han pasado a denominarse Parálisis Facial Hereditaria ya que se consideran una entidad diferente. En este contexto, el SM clásico se presenta en el 68.7%, un 25% con SM Incompleto y tan solo en el 6.3% con SM-Like.
Bibliografía
- A Pérez Aytés. Síndrome de Moebius. Protoc diagn ter pediatr. 2010;1:80-4.
- Fujita I, Koyanagi T, Kukita J, Yamashita H, Minami T, Nakano H et al. Moebius syndrome with central hypoventilation and brainstem calcification: a case report. Eur J Pediatr. 1991; 150:582-3.
- González CH, Vargas FR, Álvarez Pérez AB, Kim CA, Brunoni D, Marqués-Días MJ et al. Limb deficiency with or without Möbius sequence in seven brazilian children associated with misoprostol use in the first trimester of pregnancy. Am J Med Genet. 1993;47:59-64.
- Pastuszak AL, Schuler L, Speck-Martins CE, et al. Use of misoprostol during pregnancy and Möbius’ syndrome in infants. N Engl J Med 1998; 338:1881-1885.
- Govaert P, Vanhaesebrouck P, de Praeter C, Fränkel U, Leroy J. Moebius sequence and prenatal brainstem ischemia. Pediatrics. 1989;84: 570-3.
- Mulliken JB, Abramson DL, Cohen MM. Möbius Syndrome: Classification and Grading System. Plast Recons Surg 1998; 102(4): 961-967.
- Bouwes Bavinck JN, Weaver DD, Opitz JM, Reynolds JF. Subclavian artery supply disruption sequence: hypothesis of a vascular etiology for Poland, Klippel-Feil, and Moebius anomalies. Am J Med Genetics 1986;23(4):903-918.
- Ramos, F. 2000. Protocolos diagnósticos y terapeúticos de la A.E.P. Asociación Española de Pediatría. 29-32.
- Sepúlveda, W.1999. Trisomía 13: Diagnóstico citogenético prenatal y hallazgos ultrasonográficos. Revista Chilena de Ultrasonografía. 2 (1). 23-27.
- Legum C, Godel V, Nehmet P. Heterogeneity and pleiotropism in the Moebius syndrome. Clínicasl Genetics 1981;20(4): 254-259.
- Borbolla-Pertierra AM, Acevedo-González P., Bosch-Canto V., Ordaz-Favila JC, Juárez-Echenique JC. Manifestaciones oculares y sistémicas del síndrome de Möebius. An Pediatr (Barc), 2014;81(5): 297:302.
- Carta A., et al. Ophtalmologic and Systemic Features in Moebius Syndrome. An Italian Case Series. Ophtalmology 2011; 118(8): 1518-1523.
- Uzumcu, B. Karaman, G. Toksoy, Z.O. Uyguner, S. Candan, H. Eris, et al.
- Molecular genetic screening of MBS1 locus on chromosome 13 for microdeletions and exclusion of FGF9, GSH1 and CDX2 as causative genes in patients with Moebius syndrome.
- Eur J Med Genet., 52 (2009), pp. 315-320.
- Ramos de Souza-Dias C., et al. Further Considerations about the Ophtalmic features of the Moebius Sequence, with data of 28 cases. Arq Bras Oftalmol 2007; 70(3): 451-457.
- Ziter FA, Wiser WC, Robinson A. Three-Generation Pedigree of a Moebius syndrome variant with chromosome translocation. Arch Neurol. 1977;34: 437-442. 6. Slee JJ, Smart RD, Viljoen DL. Deletion of chromosome 13 in Moebius Syndrome. J Med Genet 1991; 28: 413-414.
- Terzis JK, Noah EM. Möbius and Móbius-like Patients: Etiology, diagnosis and treatment options. Clin Plastic Surg 2002; 29; 497-514.
- Johana Andrea Botero Hernández, Andrea Paola Camargo Rojasb, Eugenia Teresa Espinosa García. Síndrome de Moebius: manifestaciones neurológicas, musculoesqueléticas y del lenguaje. Repertorio de Medicina y Cirugía. Vol. 26. Núm. 2. páginas 109-112 (Abril – Junio 2017)
- Pablo Arrieta-Joffe, Marcia Pérez-Dosal, gabriela ortiz-de-zarate, alexander cárdenas-mejía. Estudio clínico, citogenético, molecular y de imagen de los pacientes con síndrome de Moebius del Hospital General “Dr. Manuel Gea González”, Ciudad de México Clínicasl, cytogenetic, molecular and image study of patients with Mobius syndrome at the General Hospital “Dr. Manuel Gea González”, Mexico city. Cir. plást. iberolatinoam.-Vol. 43 – Nº 4 Octubre – Noviembre – Diciembre 2017 / Pag. 395-400 http://dx.doi.org/10.4321/S0376-78922017000500010
- Calderón León, María; Calle Morillo, Lennys; Hidalgo Acosta, Javier. Reporte de caso clínico: síndrome de Moebius asociado al uso de misoprostol en el embarazo / Clínicasl case report: Moebius syndrome associated with the use of misoprostol in pregnancy. Medicina (Guayaquil) ; 17(1): 65-69, mayo 2012.