INFORMACIÓN BÁSICA.- Síndrome de McCune-Albright. (OMIM 174800/ORPHAN:/ CIE-10: ).- El síndrome de McCune-Albright (SMA) se define clásicamente por la tríada clínica de displasia fibrosa ósea (DF), manchas cutáneas café con leche y pubertad precoz (PP).
La DF puede involucrar a uno o varios sitios del esqueleto y se presenta con cojera y / o dolor y, ocasionalmente, una fractura patológica. La escoliosis es común y puede ser progresiva. Además de PP (sangrado vaginal o manchado y desarrollo de tejido mamario en niñas, aumento testicular y peneano y comportamiento sexual precoz en niños), otras endocrinopatías hiperfuncionantes pueden estar involucradas incluyendo hipertiroidismo, exceso de hormona de crecimiento, síndrome de Cushing y pérdida renal de fosfato.
Las manchas Café con leche los puntos generalmente aparecen en el período neonatal, pero lo más frecuente es que PP o DF atraigan al niño a atención médica. La afectación renal se observa en aproximadamente el 50% de los pacientes con SMA. La enfermedad es el resultado de mutaciones somáticas del gen GNAS, específicamente mutaciones en la proteína reguladora cAMP, G s alfa. La extensión de la enfermedad está determinada por la proliferación, migración y supervivencia de la célula en la que la mutación ocurre espontáneamente durante el desarrollo embrionario. El diagnóstico de SMA generalmente se establece sobre bases clínicas. Las radiografías simples a menudo son suficientes para hacer el diagnóstico de DF y la biopsia de las lesiones DF puede confirmar el diagnóstico. La evaluación de los pacientes con SMA debe guiarse por el conocimiento del espectro de tejidos que pueden estar involucrados, con pruebas específicas para cada uno. Las pruebas genéticas son posibles, pero no están disponibles de forma rutinaria. Sin embargo, se debe ofrecer asesoramiento genético. Los diagnósticos diferenciales incluyen neurofibromatosis, displasia osteofibrosa, fibromas no osificantes, pubertad precoz central idiopática y neoplasia ovárica. El tratamiento es dictado por los tejidos afectados, y la medida en que se ven afectados. En general, se recomienda alguna forma de intervención quirúrgica. Los bisfosfonatos se utilizan con frecuencia en el tratamiento de DF. Se recomiendan ejercicios de fortalecimiento para ayudar a mantener la musculatura alrededor del hueso DF y minimizar el riesgo de fractura. El tratamiento de todas las endocrinopatías es obligatorio. Las malignidades asociadas con SMA son casos claramente raros. La transformación maligna de las lesiones DF ocurre en probablemente menos del 1% de los casos de SMA. Las malignidades asociadas con SMA son casos claramente raros. La transformación maligna de las lesiones DF ocurre en probablemente menos del 1% de los casos de SMA. Las malignidades asociadas con SMA son casos claramente raros. La transformación maligna de las lesiones DF ocurre en probablemente menos del 1% de los casos de SMA.
Definición.- Originalmente, el SMA se definió por la tríada de DF poliostótica, pigmentación de la piel café con leche y PP. Más tarde se reconoció que otras endocrinopatías, incluyendo hipertiroidismo (revisado en exceso de hormona de crecimiento (GH) pérdida renal de fosfato con o sin raquitismo/osteomalacia y síndrome de Cushing podrían encontrarse en asociación con la tríada original en raras ocasiones, otros sistemas de órganos pueden estar involucrados (hígado, corazón, paratiroides, páncreas).
Mientras que SMA es raro, DF no lo es. La DF puede involucrar un solo sitio esquelético (FD monostótica, DFM), o múltiples sitios (DF poliostótica, DFP) .Muy rara vez se puede encontrar PP en asociación con pigmentación de la piel café con leche en ausencia de DF (aproximadamente 1% de los casos), pero en general, DF parece ser el componente más común de SMA. Por lo tanto, una definición más clínicamente relevante de SMA, más amplia que la tríada original de DF + PP + café con leche es: SMA = DF + al menos una de las típicas endocrinopatías hiperfuncionantes y / o manchas de café con leche, con casi cualquier combinación posible.
Epidemiología.- SMA es una enfermedad rara y no se dispone de datos confiables de prevalencia (la prevalencia estimada oscila entre 1/100,000 y 1/1,000,000). Por el contrario, el aspecto esquelético de la enfermedad, DF, especialmente la enfermedad monostótica, no es raro. Se ha informado que DF representa hasta el 7% de todos los tumores óseos benignos.
Descripción clínica.- Típicamente, los signos y síntomas de PP o FD generalmente explican la presentación inicial. En las niñas con PP, generalmente es un sangrado vaginal o manchado, acompañado por el desarrollo de tejido mamario, generalmente sin desarrollo de vello púbico. En los niños, puede ser agrandamiento testicular bilateral (o unilateral) con agrandamiento del pene, ruga escrotal, olor corporal, vello púbico y axilar y comportamiento sexual precoz.
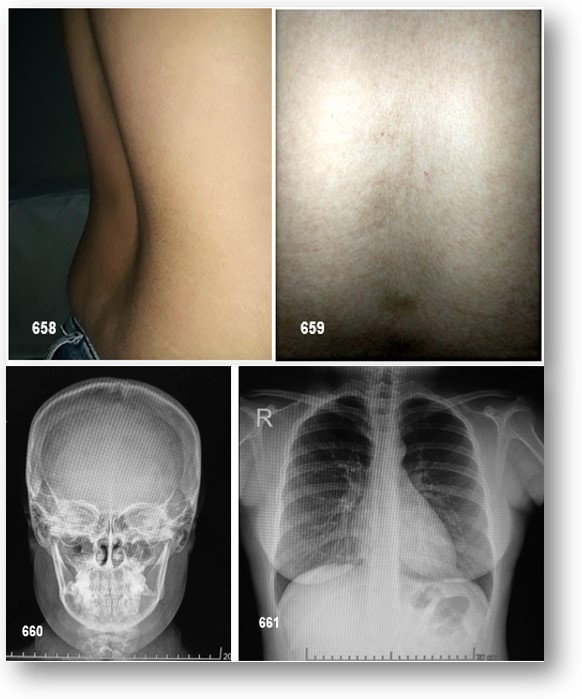
En retrospectiva, los puntos de café con leche (Imagen 659), que suelen estar presentes al nacer o poco después, son los signos más comunes pero no apreciados de «presentación».
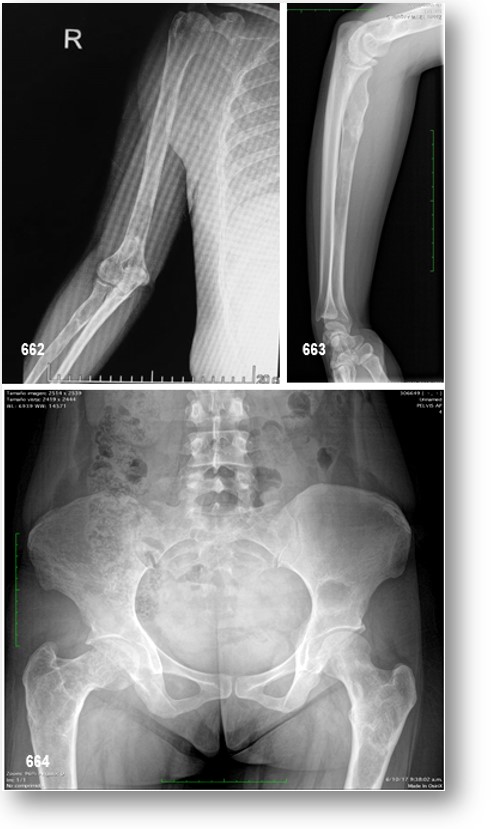
La DFP puede afectar al 75% del esqueleto. Las lesiones óseas generalmente son asintomáticas y su crecimiento es lento y progresivo, lo que provoca asimetría. Su etiología no es bien conocida, pero algunos estudios indican un trastorno congénito con una actividad aberrante del tejido mesenquimal anterior del hueso o una interrupción de la maduración ósea en la fase de desarrollo. La DF en el esqueleto apendicular por lo general se presenta con cojera y / o dolor (a veces informado por los niños como «cansado»), pero ocasionalmente una fractura patológica puede ser el signo de presentación. Las radiografías mostrarán lesiones típicas expansibles con festoneo endostal y adelgazamiento de la corteza con la matriz del tejido intramedular demostrando una apariencia de «vidrio esmerilado» (Figura 65-C). La DF en los huesos craneofaciales por lo general se presenta como un «bulto» o asimetría facial indolora. Las áreas más comúnmente involucradas son el fémur proximal y la base del cráneo. Los sitios de participación de DF se establecen temprano.
Otras características de la presentación relacionadas con el aspecto específico de la enfermedad se describen en la tabla 54.
El 90% de la carga total de enfermedad del esqueleto corporal generalmente se establece a los 15 años. Hart et al. encontraron que las lesiones en la región craneofacial se establecieron antes, con el 90% de las lesiones presentes a los 3.4 años de edad. En las extremidades, el 90% estaba presente por 13.7 años, y en el esqueleto axial, el 90% estaba presente por 15.5 años.
La aparición de nuevas lesiones más adelante en la vida es una ocurrencia muy poco frecuente en DF. La incidencia de fracturas es mayor en la infancia, entre los 6 y 10 años, pero debido a las anomalías intrínsecas en el hueso DF, algunas fracturas continúan ocurriendo hasta la edad adulta.
Pubertad precoz.- La pubertad precoz y la displasia ósea fibrosa son las alteraciones más frecuentes en el síndrome de McCune-Albright. La PPP se produce por hiperfunción gonadal autónoma, con manifestaciones clínicas características, diferentes de la PPC. Mientras en la PPC la edad de comienzo generalmente es cercana a la de la pubertad normal y la progresión del desarrollo semejante, en la PPP la edad del primer signo es muy temprano, habitualmente se manifiesta por sangrado vaginal, que puede preceder o ser simultáneo al inicio de la telarca, y hay escasa aceleración de la velocidad de crecimiento y de la edad ósea, como consecuencia de la rapidez con que se instala el cuadro. La demostración ecográfica de quistes ováricos, que involucionan espontáneamente, y su reaparición acompañada de los signos clínicos en forma reiterada orientan al diagnóstico, como sucedió en todas nuestras pacientes. La confirmación del carácter periférico de la PP se efectúa mediante determinación de gonadotrofinas basales o bajo estímulo con GnRH, que muestran valores bajos o inhibidos, y estradiol sérico cuyos resultados son variables, como ocurrió en este estudio. Cuando los quistes ováricos involucionan, los niveles de estradiol caen rápidamente, mientras que la inhibición de las gonadotrofinas puede persistir, de ahí la discordancia registrada en algunos casos. La PPP suele presentarse con evolución intermitente y largos períodos de remisión, sin embargo, en algunos casos se observa progresión rápida, con aceleración del crecimiento y la maduración esquelética, que comprometen la talla final. Como consecuencia de la exposición sostenida o reiterada a los niveles elevados de estradiol secretados por los quistes foliculares, puede desarrollarse secundariamente PPC por reactivación prematura del eje HHG, lo que se observó en 2 niñas en esta serie. El diagnóstico de que la PPP corresponde a un SMA es clínico cuando se presentan las tres manifestaciones clásicas. En los casos de ausencia aparente de alteraciones óseas, está indicado efectuar un centellograma óseo con Tc99, que se hizo en todas nuestras pacientes; y si se detectan áreas de hipercaptación, efectuar radiografía localizada en la zona para precisar la lesión. Si hay dudas diagnósticas, se recurre al estudio molecular para detectar la mutación, que en esta serie de pacientes no fue posible realizar en todos los casos. Cuando el análisis se efectúa en un tejido afectado, la mutación se encuentra en más del 90 % de los casos, independientemente de la presentación clínica. En cambio, en muestras de sangre periférica, se detecta en una proporción mucho menor, y menor aún si la paciente no presenta la tríada clásica del SMA. Se han utilizado diferentes tratamientos para tratar de disminuir la formación de quistes ováricos y los efectos del hiperestrogenismo, con diferente eficacia. Si bien el AMP puede inhibir los sangrados vaginales, no está demostrado que tenga efecto sobre la maduración ósea.
El tratamiento con tamoxifeno, modulador selectivo del receptor de estrógenos, en 25 niñas, mostró disminución de los episodios de SV y buen control de la velocidad de crecimiento y la maduración ósea, aunque no tuvo efecto sobre el volumen ovárico y el tamaño uterino. Otro estudio en 8 niñas tratadas entre 3 y 8 años logró el cese del SV, la estabilización de la maduración esquelética y mejoró la predicción de la talla final. El letrozole, inhibidor de aromatasa de tercera generación, se considera una terapéutica útil en la PPP. Un estudio en 9 niñas disminuyó la tasa de SV, la velocidad de crecimiento y el avance de EO. Solo 1 paciente desarrolló un quiste ovárico bajo tratamiento.26 Estrada y col., publicaron un estudio retrospectivo a largo plazo con letrozole en 28 niñas. Fue efectivo para disminuir la velocidad de crecimiento, la tasa de maduración esquelética y para mejorar el pronóstico de talla adulta. En las niñas con respuesta inadecuada al tratamiento médico, la resección quirúrgica de los quistes ováricos persistentes por quistectomía laparoscópica puede ayudar a controlar la PPP. Todas nuestras pacientes recibieron AMP por ser la única terapéutica accesible hasta el año 1987; tuvieron respuesta favorable con respecto al número de SV, probablemente por la acción antiestrogénica de esta progestina sobre el endometrio, pero no evidenciaron un freno en la aceleración de la edad ósea. Con la nueva terapéutica implementada en nuestra serie, no se pueden sacar conclusiones debido al reducido número de pacientes tratadas.
Existe escasa información sobre la evolución de la función ovárica en la adolescencia y en la edad adulta. Como se observó en nuestras pacientes, pueden persistir episodios de autonomía ovárica que se manifiestan por trastornos del ciclo, desarrollo de quistes y niveles elevados de estradiol con gonadotrofinas prepuberales o inhibidas.29,30 Sotomayor y col. encontraron una proporción de ciclos ovulatorios significativamente menor en las pacientes con SMA comparadas con controles normales.
La DOF suele ser unilateral y generalmente poliostótica. La base del cráneo y los huesos largos son los sitios más frecuentemente involucrados. Los estudios radiográficos muestran lesiones de aspecto quístico o lítico, y en niños son clásicas las lesiones en “vidrio esmerilado”. También pueden observarse fracturas patológicas y deformación, como ocurre en el fémur (“bastón de pastor”). Las manifestaciones iniciales son dolor, claudicación de una extremidad o una fractura patológica. La incidencia es mayor entre los 5 y 10 años. Se ha utilizado tratamiento con pamidronato en casos de dolor intenso, lo que se hizo en 4 pacientes por vía oral y 2 lo recibieron intravenoso. En huesos largos, se recomienda el tratamiento quirúrgico con colocación de prótesis intramedulares. Todas nuestras pacientes presentaron DOF; la mayoría de ellas tuvieron fracturas y un número elevado requirió cirugía. A nivel craneofacial aparecen protuberancias o asimetría facial; el exceso de GH se asocia con displasia fibrosa en la base del cráneo y con disminución de la agudeza visual y auditiva por compresión de pares craneanos.
Las manchas cutáneas color “café con leche” son un signo muy común, característico del SMA, que se observa desde el nacimiento, aunque no se les asigna valor patológico hasta que aparecen los otros elementos del síndrome, como sucedió en nuestras pacientes, que consultaron por signos de PPP. Se deben a una proliferación de los melanocitos, son benignas, sin potencial de progresión a malignidad, de número y morfología variables, casi siempre tienen bordes irregulares (en “costa de Maine”) y se hacen más evidentes con la exposición a la luz solar. Están limitadas a un hemicuerpo, generalmente no sobrepasan la línea media y no varían sus características durante la evolución de la enfermedad, como en nuestras pacientes. Se distribuyen siguiendo las líneas de Blaschko, que representan el patrón de migración de las células del ectodermo durante la etapa embrionaria.
La pubertad precoz periférica asociada al síndrome de McCune-Albright es una entidad clínica poco frecuente. Su diagnóstico se establece por lo general en la infancia, pero la aparición de los otros elementos del síndrome, como la displasia ósea y otras hiperfunciones endocrinas, suele ser más tardía y progresiva, por lo que es necesario que estas pacientes cuenten con una vigilancia clínica constante.
Malignidades en SMA
Si bien las malignidades asociadas con SMA son casos excepcionalmente raros, merecen mención debido a su importancia. La transformación maligna de las lesiones DF es probablemente la neoplasia maligna más común y mejor descrita que se produce en asociación con SMA. Esto ocurre en probablemente menos del 1% de los casos de DF/SMA. La radiación de haz externo de alta dosis es un factor de riesgo para la transformación sarcomatosa. Puede haber una mayor tendencia a que se produzca una transformación maligna en pacientes que tienen un exceso concomitante de GH.
Mientras que algunos han sugerido que la transformación sarcomatosa de las lesiones esqueléticas puede ocurrir más comúnmente en pacientes con síndrome de Mazabraud (mixomas intramusculares benignos en asociación con DF de larga duración), esto puede representar un sesgo de selección.
Además, el riesgo de cáncer de mama puede ser elevado en pacientes con SMA. Además de los informes publicados, en la serie de aproximadamente 120 pacientes atendidos en los Institutos Nacionales de Salud (NIH), la prevalencia de cáncer de mama fue de aproximadamente 2.5% (n=3). En este grupo de tres pacientes, también parece existir un efecto del exceso de hormona del crecimiento (HC), ya que todas las pacientes que tenían cáncer de mama también tenían exceso de HC (datos no publicados).
El cáncer de tiroides y el cáncer testicular (datos no publicados) también son casos raros.
Etiología.- La observación de que la vía de señalización de la proteína G/cAMP/adenilato ciclasa era fundamental para todos los tejidos implicados en el SMA finalmente condujo al descubrimiento de que las mutaciones en la proteína reguladora G s α (codificada por el gen GNAS) eran la etiología molecular subyacente de SMA (Figura 1). En todos los casos publicados de SMA, DFP e incluso DFM, se han identificado mutaciones activadoras de G s α en la posición R201. Más recientemente, se han encontrado mutaciones en la posición Q227 en asociación con DF.
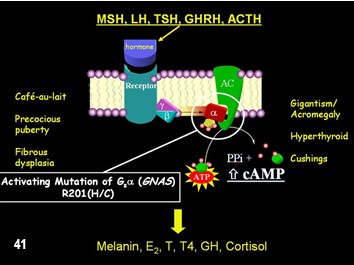
La falta de transmisión vertical de la enfermedad, junto con la observación de que las lesiones cutáneas y óseas tienden a respetar la línea media y estar en uno u otro lado del cuerpo, ha llevado al concepto no probado, pero aceptado, de que la enfermedad es la resultado de mutaciones postzigóticas, y que los pacientes son, por lo tanto, mosaicos somáticos. El punto en el tiempo en el desarrollo en el que se produce la mutación, la célula específica en la que se produce y hacia donde migra su progenie, determina qué tejidos se verán afectados, y por lo tanto el fenotipo. Por lo tanto, en los casos en los que están implicados tejidos de origen endodérmico, mesodérmico y ectodérmico, parecería que la mutación se produjo en la etapa de masa celular interna.
Diagnóstico.- Criterios de diagnóstico, métodos de diagnóstico, diagnóstico diferencial.-
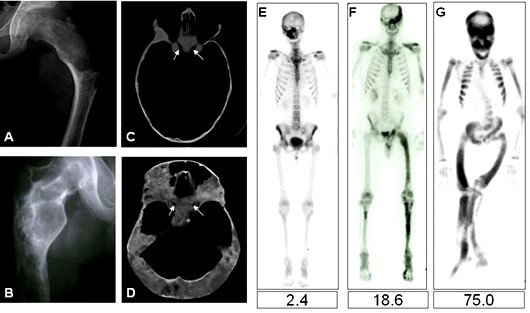
El diagnóstico de SMA generalmente se establece sobre bases clínicas. Las radiografías simples a menudo son suficientes para hacer el diagnóstico de DF. Las exploraciones óseas isotópicas son la herramienta más sensible para detectar la presencia de lesiones DF, y a menudo son útiles, especialmente en la evaluación inicial, para determinar el alcance de la enfermedad y predecir el resultado funcional.
La DF tiene una apariencia típica en las radiografías descritas como «vidrio esmerilado». En general, las lesiones en los huesos largos tienen una apariencia destructiva («lítica»). Las lesiones generalmente surgen en la cavidad medular y se expanden hacia afuera reemplazando el hueso normal, lo que produce un adelgazamiento de la corteza. Por lo general, la metáfisis y / o la diáfisis están involucradas, con preservación de la epífisis. Es posible que participe cualquier hueso, pero la base del cráneo y el fémur proximal son los sitios más comúnmente involucrados. Debido al hecho de que estas lesiones están menos mineralizadas, los huesos son «blandos» y propensos a la deformación, como se ejemplifica en la deformidad clásica del «fémur del pastor» del fémur proximal.
La DF en los huesos craneofaciales tiende a tener una apariencia «esclerótica» en radiografías simples. Esto se debe al grado relativamente mayor de mineralización del tejido DF en los huesos craneofaciales. La exploración por TAC es la mejor técnica para obtener imágenes de lesiones DF en el cráneo, revelando una apariencia de «vidrio esmerilado». En niños y adultos jóvenes, las lesiones parecen homogéneas en la TAC, pero en pacientes mayores la apariencia es mixta, con el desarrollo de lesiones «quísticas» en algunas áreas. La densidad de estas áreas es la de los tejidos blandos, por lo que si bien pueden tener una apariencia quística, no son verdaderos quistes. Dicho esto, es posible que se desarrollen quistes verdaderos en DF, tanto en los huesos largos como con mayor frecuencia en los huesos craneofaciales. Esto ha ocurrido en aproximadamente el 5% de los pacientes con DF en la cohorte NIH (datos no publicados). Si es necesario, los quistes óseos pueden diagnosticarse mediante RMN. Los quistes tienden a tener un curso más agresivo. Se pueden expandir rápidamente y producir síntomas que varían según la ubicación. Una de las complicaciones puede ser la fractura a través de un quiste. Esto usualmente requiere intervención quirúrgica.
Prueba genética.- Las pruebas genéticas son posibles, pero no están disponibles de forma rutinaria. Debido a la naturaleza del mosaico somático de la enfermedad, un resultado negativo del tejido disponible (pero no afectado) no excluye la presencia de la mutación. Las pruebas de ADN de leucocitos son posibles, pero no es confiable. En la mayoría de los casos, las pruebas genéticas contribuyen poco al diagnóstico y no a la gerencia. No se conoce una correlación genotipo/fenotipo, por lo que el conocimiento de la mutación específica no afecta el tratamiento. Por esta razón, cuando se sospecha o se establece el diagnóstico de SMA, es importante conocer el espectro de tejidos que posiblemente puedan estar involucrados y detectar la afectación. La detección, como mínimo, requiere un historial médico y un examen físico, y por lo general incluye imágenes y pruebas bioquímicas específicas.
Los siguientes se enumeran como posibles fuentes de pruebas genéticas: Centro de Pruebas Genéticas en Saint Francis Genome Diagnostics. Además, los laboratorios de investigación de los profesores Francis Glorieux, Shriners Hospital for Children (Montreal, Canadá), Paolo Bianco (Roma, Italia) y Charles Sultan (Montpellier, Francia) pueden realizar pruebas genéticas con fines de investigación.
Pruebas complementarias:
– La determinación de gonadotropinas basales y tras estímulo con LHRH (100μg/m2 iv) es primordial para el diagnóstico diferencial entre Pubertad Precoz Central (PPC) y Pubertad Precoz Periférica (PPP). Si bien no existe unanimidad en la comunidad científica internacional sobre el punto de corte de LH para definir PPC y PPP, la mayoría de autores lo sitúan entre 5 y 8 U/l3.
Además del estudio de gonadotropinas basales y tras estímulo, existen otras determinaciones sanguíneas que servirán de complemento en el diagnóstico diferencial de PPP; son las que siguen:
- – Testosterona: sus niveles séricos son de utilidad para el diagnóstico de pubertad precoz en el niño. Así, valores por encima de 0,5ng/ml se consideran en rango puberal.
- – 17-β-estradiol: escasa sensibilidad, ya que valores normales, no descartan pubertad precoz; sin embargo, se encuentra muy elevada en presencia de tumores ováricos y suprarrenales productores de estrógenos, así como ante quistes ováricos aislados o asociados a SMA.
- – DHEA-S, androstendiona (Δ4) y 17-OH-progesterona: útiles en el niño con sospecha de pubertad precoz. Si encontramos cifras de DHEA-S por encima de 700μg/dl en un niño en edad prepuberal, es altamente sugerente de tumor suprarrenal. Elevaciones moderadas, tras prueba de imagen normal, nos obligan a descartar una hiperplasia suprarrenal de presentación tardía.
- – β-HCG (fracción β de gonadotropina coriónica humana): de utilidad como marcador tumoral en casos de pubertad precoz periférica con tamaño testicular menor de 4ml en el que no se encuentra alteración testicular ni suprarrenal (cuando se trata de tumores germinales extragonadales: hígado, mediastino, cerebro).
- Los tumores germinales testiculares productores de β-HCG producen asimetría testicular (a veces tamaño testicular > 4ml).
- – T4 libre y TSH: para descartar hipotiroidismo primario, causa excepcional de pubertad precoz.
- Por otra parte, disponemos de una serie de pruebas de imagen de enorme utilidad para el diagnóstico diferencial de las distintas formas de pubertad precoz.
- – Cálculo de la edad ósea mediante radiografía de mano-muñeca izquierda: en los casos de pubertad precoz hay paralelamente aceleración de la edad ósea, en comparación con las variantes de la normalidad. No obstante, en casos de reciente diagnóstico de PPP, la edad ósea puede ser similar a la edad cronológica.
- – Ecografía testicular: se realizará siempre que haya sospecha de pubertad precoz en el varón que presenta asimetría testicular. Asimismo en situaciones de sospecha de pubertad precoz con elevación de testosterona y volumen testicular inferior a 4ml.
- – Ecografía abdominal: de utilidad para valorar área suprarrenal, tanto en sospecha de tumores virilizantes en el varón, como en niñas con sospecha de pubertad precoz periférica por un tumor productor de estrógenos. Asimismo se realizará cuando haya indicios de tumor hepático productor de β-HCG.
- – Ecografía pélvica: se realizará en todos los casos de pubertad precoz. Por un lado, permite descartar patología causante de PPP (tumores ováricos, quistes ováricos) y, por otro, valora la existencia de signos de impregnación estrogénica.
- – RM craneal: de obligada realización en todos los casos de PPC, tanto en niños como niñas. También se efectuará en casos de PPP en el niño por sospecha de tumor productor de β-HCG extragonadal no hepático.
- – Serie ósea: para excluir la presencia de displasia fibrosa asociada a SMA.
Finalmente, ante una sospecha clínica elevada, procederemos al estudio genético:
- – Estudio del gen del receptor de LH (LHR) en la testotoxicosis.
- – Estudio del gen DAX1 ante la sospecha de una hipoplasia suprarrenal congénita.
- – Estudio gen la subunidad α de la proteína G: la rentabilidad en sangre periférica es modesta. Por lo que es de interés efectuar el estudio en tejidos afectos (ovario, hueso).
Diagnóstico diferencial.- El SMA se confunde con la neurofibromatosis (NF Tipo 1), generalmente cuando un niño presenta un gran lugar de café con leche. La ubicación y la forma de las manchas generalmente pueden ayudar a distinguir entre el SMA y el NF. Las manchas en SMA tienen bordes dentados (costa de Maine), mientras que las manchas en NF son lisas (costa de California). Aunque las manchas pueden cruzar la línea media, con mayor frecuencia, demuestran un «respeto» a la línea media. Las localizaciones frecuentes son la nuca y el pliegue en el ápice de las nalgas. En SMA, la enfermedad esquelética (DFP) casi siempre involucra uno o ambos fémures proximales y/o la base del cráneo, así como otros lugares. La afectación esquelética en la NF es poco común y generalmente involucra las diáfisis de los huesos largos, especialmente las tibias, que a menudo conducen a una seudoartrosis.
Cuando la PP es el signo de presentación, el diagnóstico diferencial incluye la pubertad precoz central idiopática y una neoplasia ovárica. Las gonadotropinas suprimidas excluyen el PP central. Otros componentes del SMA (pigmentación de la piel, cambios esqueléticos en los rayos X o gammagrafía ósea, etc.) pueden ayudar a asegurar al clínico que el quiste ovárico no es neoplásico.
Las lesiones esqueléticas aisladas en ausencia de lesiones de la piel o hallazgos endocrinos representan la DF (en sus formas, DFM o DFP). La displasia osteofibrosa (fibroma osificante de los huesos largos, llamada lesión de Campanacci) se puede confundir con DF. Estas lesiones se encuentran casi exclusivamente en la tibia y el peroné, y son histológicamente distintas de la DF. Los fibromas no osificantes (NOF) también pueden compartir similitudes radiológicas e histológicas con DF en los huesos largos. La falta de focos esqueléticos múltiples y la ausencia de hallazgos extraesqueléticos pueden ayudar a distinguir DF de NOF. DF en la mandíbula puede compartir varias características histológicas con fibromas cemento osificante, que se pueden confundir con DF. Las lesiones de fibroma cemento osificante tienden a comportarse de forma más agresiva que las lesiones DF. Prueba para el GNASLa mutación, si los tejidos y los ensayos están disponibles, pueden ser útiles para distinguir las lesiones de fibrosis cemento osificante de DF.
De presentarse el SMA con la forma de DFP, esta comparte características con muchas otras enfermedades. Por lo tanto, un diagnóstico diferencial preciso es obligatorio para establecer un enfoque de tratamiento oportuno y apropiado. El diagnóstico diferencial debe basarse en hallazgos clínicos, radiográficos e histopatológicos y debe incluir lesiones periapicales, fibroma osificante, enfermedad de
Paget, osteomielitis crónica, neurofibromatosis, querubismo, granuloma de células gigantes, quiste óseo aneurismático, cemento -displasia florosa -oseosa, histiocitosis X, mieloma múltiple, osteosarcomas, fibrosarcomas y condrosarcomas.
Consideraciones diagnósticas específicas y seguimiento
Esqueleto.- La enfermedad esquelética, especialmente la que afecta a la base del cráneo, es muy común. La pérdida de visión y/o audición es poco común y la transformación sarcomatosa es rara (<1%). DF en el esqueleto apendicular tiende a calmarse con la edad, pero el curso de la enfermedad craneofacial es menos claro. Las exploraciones óseas de medicina nuclear son útiles no solo para detectar el alcance de la enfermedad, sino también para cuantificar la carga de la enfermedad esquelética de DF y predecir el resultado funcional.
DF craneofacial.- La tomografía computarizada del cráneo es la prueba más útil para el diagnóstico de DF craneofacial. La TC debe repetirse periódicamente, anualmente y luego con menos frecuencia una vez que se ha demostrado la estabilidad. La visión debe ser evaluada, idealmente por un neurooftalmólogo inicialmente, luego periódicamente; anualmente o con menos frecuencia una vez que se ha demostrado la estabilidad. La evaluación auditiva se recomienda al inicio del estudio para evaluar la participación y también debe repetirse periódicamente. Todas las endocrinopatías que afectan negativamente al hueso deben ser examinadas y tratadas. El exceso de HC en particular puede afectar adversamente la DF craneofacial 99 Tc-metil difosfonato (99Tc-MDP) exploración ósea en la línea de base y periódicamente se recomienda.
Esqueleto axial y apendicular.- Las radiografías simples, que demuestran la apariencia clásica del vidrio esmerilado, suelen ser suficientes para el diagnóstico de DF en el esqueleto axial y apendicular (el esqueleto axial está compuesto por huesos del cráneo, hueso hioides, vértebra, esternón y costillas; el esqueleto apendicular incluye el cadera, hueso pélvico y la cintura escapular (clavícula y escápula). Sin embargo, los rayos X a menudo no pueden detectar microfracturas nuevas y pequeñas. Cuando se desarrolla un nuevo dolor focal en una lesión DF y no hay fractura evidente en el plano La radiografía, la TC o la RM pueden ser útiles para detectar fracturas sutiles. La lesión clásica del fémur proximal en DF, la deformidad del ladrón del pastor es común.
Endócrino: Gónadas.- El PP es más común en las niñas que en los niños. Sin embargo, pequeñas masas testiculares de Leydig y / o hiperplasia de células de Sertoli son comunes en los niños. Por lo general, nunca se detectan en el examen físico. Por lo tanto, se recomienda la ecografía de los testículos en todos los hombres con DF y SMA. Probablemente sea seguro monitorear las masas testiculares sin intervención quirúrgica ya que el cáncer es probablemente bastante raro. El PP de SMA en las niñas se debe a niveles elevados de estradiol en suero debido a la activación intermitente y autónoma del tejido ovárico. Algunos de ellos progresarán a PP central, secundarios a la activación del eje hipotalámico-pituitario-gonadal (HPG). Como el PP central generalmente se desarrolla en niños que han tenido PP durante algunos años, los niños con PP deben ser evaluados para PP central secundario.
Tiroides.- El hipertiroidismo es frecuente (38%). La evidencia de compromiso tiroideo sin hipertiroidismo franco es aún más común (63% en la serie NIH, datos no publicados). Esto se manifiesta como una hormona estimulante tiroidea (TSH) relativamente suprimida con Triiodotironina elevada (T3 +) y una glándula tiroides anormal en el ultrasonido. Algunos de estos pacientes pueden desarrollar hipertiroidismo franco, por lo tanto, seguimiento con medición de TSH, tiroxina libre (FT4), T3 y tiroxina (T4), y se recomienda la ecografía periódica para detectar aquellos pacientes que progresan a hipertiroidismo franco. Si bien el desarrollo de cáncer de tiroides en pacientes con SMA es raro, los pacientes deben ser monitoreados por esta posibilidad. Esto se logra mediante una ecografía anual de la tiroides. La apariencia ecográfica de la tiroides en el SAM es complicada. Por lo general, hay áreas mezcladas de tiroides de apariencia relativamente normal con áreas quísticas adyacentes. La sospecha de cáncer se debe elevar si se observa una lesión sólida grande o en expansión. Esto debería impulsar la punción aspiración con aguja fina (PAAF) con evaluación citopatológica.
Pérdida de fosfato renal.- La pérdida renal de fosfato, como parte de una tubulopatía proximal, con o sin hipofosfatemia y/o raquitismo/osteomalacia es común. La etiología es probable debido a la elaboración del factor fosfatúrico, factor de crecimiento de fibroblastos-23 (FGF23), por tejido DF.
Se recomienda la medición del fosfato sérico y el cálculo del manejo del fosfato renal mediante la reabsorción tubular de fosfato (TRP) o la reabsorción tubular máxima de fosfato por tasa de filtración glomerular (TmP/GFR). Si bien es discutible si hipofosfatemia, per se, debe tratarse o si el tratamiento debe reservarse solo para pacientes con raquitismo o raquitismo demostrado por la biopsia, es nuestra sensación de que la franca hipofosfatemia debe ser tratada.
Hipófisis.- El exceso de HC y prolactina es común (21%) y los signos y síntomas pueden ser muy sutiles. Dado que el exceso de HC puede empeorar la enfermedad ósea craneofacial, es importante hacer el diagnóstico y el tratamiento. Todos los pacientes deben tener una prueba de tolerancia oral a la glucosa (TOG) para buscar HC no suprimible al menos una vez. La mayoría de los tumores que secretan HC son co-secretores de HC y prolactina, por lo que los niveles de prolactina también deben evaluarse, ya que la hiperprolactinemia puede tener un efecto adverso independiente en la función gonadal.
Paratiroides.- El hiperparatiroidismo primario en el SMA es raro y probablemente no forma parte del síndrome. El hiperparatiroidismo secundario, generalmente debido a la deficiencia de vitamina D, es común en la población general así como en DF/SMA. El hiperparatiroidismo puede empeorar la DF y debe tratarse. El calcio sérico total o ionizado y la hormona paratiroidea (PTH) deben evaluarse periódicamente.
Suprarrenal.- El síndrome de Cushing puede ocurrir en el período neonatal, pero no se ha informado después del primer año. Algunos casos de Cushing neonatal se resuelven espontáneamente. El síndrome de Cushing debe ser considerado y evaluado en pacientes muy jóvenes. El examen físico suele ser sugestivo y muestra facies lunar con plétora, hirsutismo y falta de crecimiento lineal. Si se sospecha, se recomienda una evaluación de laboratorio con una combinación de orina de 24 horas para cortisol urinario libre (si es posible), medianoche o cortisol salival, y/o prueba de supresión con dexametasona. Se debe controlar la reserva suprarrenal en pacientes en quienes Cushing se resolvió espontáneamente (comentario personal, en la serie NIH 2 de cada 9 pacientes con Cushing tenían curación espontánea y reserva suprarrenal inadecuada más adelante en la vida).
Asesoramiento genético.- El aspecto más importante de aconsejar a las familias es asegurar a los pacientes o las familias que no hay transmisión vertical de la enfermedad, ni existen asociaciones ambientales conocidas o predilección por grupos étnicos. Por lo tanto, los padres no necesitan sentirse «responsables», y los pacientes pueden estar seguros de que no transmitirán la enfermedad a sus hijos.
Diagnóstico prenatal.- Irrelevante.
Manejo, tratamiento y pronóstico
- Esqueleto
- FD craneofacial
En la gran mayoría de los casos de DF craneofacial, la cirugía no es necesaria, y la observación es el enfoque correcto. La observación involucra pruebas anuales de visión y audición e imágenes. La visión debe ser revisada por un neurooftalmólogo. La TC de cráneo y/o mandíbula es la prueba de imagen de elección para las estructuras craneales en general, pero la IRM proporcionará una mejor resolución de los nervios ópticos. Los nervios craneales a menudo están rodeados por hueso DF, pero no se ven afectados durante años o décadas. Las indicaciones para la cirugía incluyen alteración visual progresiva documentada, dolor intenso o desfiguración grave. La cirugía debe evitarse en ausencia de deterioro visual o auditivo. Sin embargo, cuando la cirugía está indicada, es muy importante encontrar un equipo craneofacial y neuroquirúrgico con experiencia en el tratamiento de la DF craneofacial. Es posible que la enfermedad craneofacial continúe progresando lentamente hasta la edad adulta, pero este tema no ha sido bien estudiado de manera prospectiva.
Hay poca evidencia de que los bisfosfonatos sean efectivos en la DF craneofacial, incluso para el dolor, pero en el caso de dolor que es difícil de manejar, se debe considerar el tratamiento con bisfosfonatos.
Esqueleto axial y apendicular.- El manejo quirúrgico de DF ha evolucionado a lo largo de los años. Mientras que el curetaje y el injerto óseo se consideraron una vez el tratamiento de elección, los expertos en el cuidado de la DF ya no sienten que esto sea efectivo. Los dispositivos intramedulares son preferibles, en general. Los arriostramientos pueden ser útiles, pero no han sido bien estudiados. La deformidad del ladrón de pastor puede desarrollarse, a veces muy rápido, y la gestión en niños en crecimiento es muy desafiante. En general, se recomienda alguna forma de intervención quirúrgica. Se deben realizar exámenes de detección y tratamiento de todas las endocrinopatías que afectan negativamente al esqueleto. Esto incluye PP, hipertiroidismo, síndrome de Cushing, exceso de HC e hiperparatiroidismo secundario.
Los bisfosfonatos de primera generación como el etidronato y clodronato (Bonefos®), de segunda generación como el pamidronato (Aminomux®), alendronato (Marvil 70®) y el tiludronato (Tildren®), y los de tercera generación como el risedronato (Oxidren®), zolendronato (Zolnic®) y el ibandronato (Idena®), se utilizan con frecuencia en el tratamiento de DF. El pensamiento original en el uso de los bifosfonatos en DF fue que evitarían la progresión de la enfermedad. Sin embargo, trabajos más recientes concluyen que los bisfosfonatos tienen ningún efecto sobre la historia natural de la enfermedad. Está claro, sin embargo, que los bisfosfonatos son efectivos para aliviar el dolor óseo asociado con DF. Se han sugerido muchos regímenes. Se desconoce la seguridad a largo plazo de los bisfosfonatos, especialmente en niños, y los efectos de dosis relativamente altas de bisfosfonatos en el hueso normal no afectado por DF. Por lo tanto, para minimizar la exposición, el enfoque ha sido utilizar la dosis mínima con el intervalo más largo posible entre las dosis necesarias para lograr y mantener el alivio del dolor.
Mantener la musculatura alrededor del hueso con DF es importante para protegerlo. Por lo tanto, los ejercicios de fortalecimiento y el mantenimiento de la fuerza son importantes. La natación y el ciclismo son excelentes modos de ejercicio para los pacientes con DF que promoverán el fortalecimiento y minimizarán el riesgo de fracturas.
La escoliosis es común y puede ser progresiva. Por lo tanto, es importante detectar la presencia de escoliosis y controlar cualquier progresión. Una progresión significativa puede ocurrir dentro de un corto período de tiempo.
Endócrino.- Gónadas.- La gestión de PP en niños y niñas se detalla en el archivo adicional. La clase de fármacos con la historia más larga de seguridad y eficacia son los inhibidores de la aromatasa. Más recientemente, el antagonista/agonista de estrógenos, el tamoxifeno (Taxus®), también se ha mostrado prometedor. El PP central secundario en niños con SMA se desarrolla con frecuencia después de un período de PP. Su manifestación es a menudo la progresión de la pubertad en un niño que previamente había sido bien controlado con terapia médica. El tratamiento del PP central secundario en niños con SMA es el mismo que en los niños con PP idiopático e implica el uso de análogos de la hormona liberadora de la gonadotropina de acción prolongada.
El ultrasonido se usa para detectar y seguir a niños y hombres con masas testiculares. Estos son generalmente tumores de Leydig, pero los testículos pueden demostrar una mezcla de masas celulares de Leydig y Sertoli. La microlitiasis también se ve comúnmente. El desarrollo de malignidad en los testículos de hombres con SMA parece ser muy raro. Por lo tanto, el manejo prudente puede ser la observación con ecografías testiculares anuales y la TC del tórax, el abdomen y la pelvis si existe alguna sospecha de malignidad. Las lesiones deben seguirse anualmente con ultrasonido. No hay casos reportados de malignidad ginecológica en mujeres con SMA.
Tiroides.- Los medicamentos antitiroideos, como las tionamidas (Carbimazol/NeoMercazole®, metimazol/Tapazol® y propiltiouracilo/Tirostat 50®), suelen ser eficaces para controlar el hipertiroidismo en el SMA. La resolución espontánea del hipertiroidismo en el SMA casi nunca ocurre, por lo que finalmente se indica alguna forma de tratamiento definitivo, ya sea en forma de cirugía o radiación. Se deben realizar ecografías periódicas de la tiroides para seguir las lesiones anualmente. Si se identifica una lesión dominante o cambiante, la aspiración con aguja fina está indicada para excluir el cáncer, dado que el cáncer de tiroides se puede ver en el contexto del SMA.
Renal.- La afectación renal, en forma de pérdida renal de fosfato, se observa en aproximadamente el 50% de los pacientes con SMA. Sin embargo, solo algunos de esos pacientes tendrán un grado de pérdida de fosfato que produce hipofosfatemia (el 18% de la población total se ve en NIH). Es discutible si la hipofosfatemia garantiza tratamiento con suplementos de fósforo oral si no hay signos de raquitismo. Sin embargo, recomendamos que se trate a pacientes con fosfato sérico francamente bajo. El tratamiento es el mismo que para los pacientes con hipofosfatemia ligada a X y osteomalacia inducida por tumor, dosis alta de fosfato oral con altas dosis de calcitriol (Rocaltrol®).
Hipófisis.- Se observa exceso de HC en aproximadamente el 20% de los pacientes con SMA. Se ha evidenciado que el exceso de HC se asocia con pérdida de visión, audición y macrocefalia en pacientes con SMA. Por este motivo deben tratarse los pacientes con exceso de HC y un factor de crecimiento similar a la insulina 1 elevado (IGF-1). Las opciones de tratamiento incluyen medicamentos, cirugía y radiación.
El tratamiento médico efectivo incluye análogos de somatostatina de acción prolongada y el antagonista del receptor de HC, pegvisomant (Somavert®).
Existe una mayor eficacia y seguridad a largo plazo con los análogos de somatostatina, especialmente en niños. Pegvisomant es significativamente más caro que los análogos de somatostatina y puede estar asociado con más efectos secundarios. La meta del tratamiento en un niño en crecimiento es reducir la puntuación Z del IGF-1 sérico calculada a 0.
La cirugía es una opción, pero a veces no es posible debido al engrosamiento masivo de los huesos del cráneo. Un abordaje transesfenoidal es el método preferido en la cirugía hipofisaria, pero la base del cráneo casi siempre está involucrada con la DF en pacientes con SMA y exceso de HC, por lo que este enfoque es a menudo imposible, o difícil, en el mejor de los casos. Un abordaje trans-frontal es un abordaje menos deseable de la hipófisis, en general, y en el SMA también suele complicarse con DF. Además, en pacientes con exceso de SMA y HC, la glándula pituitaria casi siempre está involucrada difusamente con áreas de hiperplasia y adenoma. Por lo tanto, la única esperanza para «curar» el exceso de HC es una hipofisectomía total. Este puede ser el mejor tratamiento en algunos casos, pero los neurocirujanos.
La radioterapia es una opción cuando la cirugía no es posible y el tratamiento farmacológico falla. Sin embargo, puede haber un mayor riesgo de transformación sarcomatosa.
Dado que la mayoría de los tumores que secretan HC secretan conjuntamente prolactina, los agonistas dopaminérgicos llamados ergolínicos como la cabergolina (Dostinex®), bromocriptina (Parlodel®), lisurida (Dopergin®), y la pergolida (Pharken®), suelen ser necesarios y son efectivos para normalizar la prolactina sérica.
Paratiroides.- Aunque se ha informado que el hiperparatiroidismo primario es parte del síndrome en el pasado, este podría no ser el caso. Cuando las mutaciones que causan SMA se buscaron en el tejido paratiroideo, no estaban presentes. Además, es probable que la secreción e hiperplasia de la glándula paratiroidea no sea un fenómeno mediado por AMPc, G s α. El hiperparatiroidismo secundario debido a la deficiencia de vitamina D es frecuente y puede empeorar la DF. Por lo tanto, debe ser examinado y tratado.
Suprarrenal.- El síndrome de Cushing es una de las manifestaciones más raras, pero más profundas del SMA. En algunos casos, puede resolverse espontáneamente, pero en la mayoría se requiere tratamiento y, en algunos casos, a pesar de la mejor atención, puede ser fatal.
La adrenalectomía es probablemente el tratamiento de elección. Si el niño está muy enfermo, el bloqueo suprarrenal puede proporcionar estabilización de la enfermedad y ganar tiempo hasta un momento en el que se puede realizar la cirugía. Las anormalidades de la función hepática generalmente acompañan al síndrome de Cushing neonatal, por lo que el ketoconazol (Nizoral®) como terapia medicinal generalmente no es una opción. Se recomienda metirapona (Metopirone®) a una dosis inicial de 300 mg/m2/día. La dosis puede aumentarse hasta 1200 mg/m2/día. En pacientes con antecedentes de Cushing neonatal, se debe controlar la reserva suprarrenal. El síndrome de Cushing en el SMA suele ir acompañado de hipertiroidismo, que, si no se trata, puede complicar y empeorar el curso clínico.
Preguntas no resueltas.- Si bien los bisfosfonatos suelen ser eficaces para aliviar el dolor relacionado con DF, si el tratamiento de la DF con bisfosfonatos cambia o no, la historia natural de la enfermedad sigue siendo una pregunta abierta. Los datos más recientes hasta la fecha sugieren que no lo hacen. Los ensayos en curso controlados con placebo en los EE. UU. y Europa debería ayudar a resolver esta pregunta. Se necesita un tratamiento más efectivo para DF. El desarrollo de la célula basada y / o G s alphadirigido terapias puede ser prometedora.
El mejor tratamiento para PP tampoco está claro. Recientemente se ha demostrado que el letrozol (Femara®), un potente inhibidor de la aromatasa de tercera generación, es una terapia eficaz en algunas niñas con SMA. Los estudios sugieren que el tamoxifeno (Taxus®), agonista/antagonista de los estrógenos, también puede ser efectivo. Sin embargo, todos los ensayos para el tratamiento del PP han sido descontrolados, y este hecho, combinado con la extrema variabilidad en el curso clínico de la enfermedad, hace que las conclusiones sobre la eficacia relativa sean muy difíciles.
Está en marcha una prueba con el antiestrógeno puro fulvestrant (Faslodex®) en niñas con SMA. Se necesitan ensayos de comparación controlados y comparativos para establecer el mejor tratamiento para PP en el SMA.
Bibliografía
- McCune DJ: Osteitis fibrosa quística: el caso de una niña de nueve años que también exhibe pubertad precoz, pigmentación múltiple de la piel e hipertiroidismo. Am J Dis Child. 1936, 52: 743 – 744.
- Albright F, Mayordomo AM, Hampton AO, Smith P: Síndrome caracterizado por osteítis fibrosa diseminada, áreas, de pigmentación y disfunción endocrina, con pubertad precoz en las mujeres: informe de 5 casos. N Engl J Med. 1937, 216: 727-746.
- Mastorakos G, Mitsiades NS, Doufas AG, Koutras DA: Hipertiroidismo en el síndrome de McCune-Albright con una revisión de anomalías tiroideas sesenta años después del primer informe. Tiroides. 1997, 7: 433 – 439.
- Sherman SI, Ladenson PW: terapia de octreotida del exceso de la hormona de crecimiento en el síndrome de McCune-Albright. Revista de investigación endocrinológica. 1992, 15: 185-190.
- Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, Cherman N, Bianco P, Wientroub S, Robey PG, Collins MT: Caracterización del exceso de hormona de crecimiento mediada por gsp en el contexto del síndrome de McCune-Albright. J Clin Endocrinol Metab. 2002, 87 (11): 5104 – 5112. 10.1210 / jc.2001-012022.
- Collins MT, Chebli C, Jones J, Kushner H, Consugar M, Rinaldo P, Wientroub S, Bianco P, Robey PG: la pérdida de fosfato renal en la displasia fibrosa del hueso es parte de una disfunción tubular renal generalizada similar a la observada en tumor- osteomalacia inducida. J Bone Miner Res. 2001, 16: 806 – 813. 10.1359 / jbmr.2001.16.5.806.
- Danon M, Crawford JD: El síndrome de McCune-Albright. Ergeb Inn Med Kinderheilkd. 1987, 55: 81 – 115.
- Diaz A, Danon M, Crawford J: síndrome de McCune-Albright y trastornos debidos a mutaciones activadoras de GNAS1. J Pediatr Endocrinol Metab. 2007, 20: 853-880.
- Kirk JM, Brain CE, Carson DJ, Hyde JC, Grant DB: síndrome de Cushing causado por hiperplasia suprarrenal nodular en niños con síndrome de McCune-Albright. The Journal of pediatrics. 1999, 134: 789 – 792. 10.1016 / S0022-3476 (99) 70302-1.
- Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, Van Wyk JJ, Merino MJ, Feuillan PP, Spiegel AM: manifestaciones endocrinas y no endocrinas graves del síndrome de McCune-Albright asociadas con la activación de mutaciones de la proteína G estimuladora GS. J Pediatr. 1993, 123: 509 – 518. 10.1016 / S0022-3476 (05) 80943-6.
- Lichtenstein LJH: Displasia fibrosa del hueso: una afección que afecta a uno, varios o muchos huesos, los casos más severos pueden presentar una pigmentación anormal de la piel, desarrollo sexual prematuro, hipertiroidismo o incluso
- otras anomalías extraesqueléticas. Arco de ruta. 1942, 33: 777-816.
- Bianco P, Robey PG, Wientroub S: displasia fibrosa. Hueso pediátrico: biología y enfermedad. Editado por: Glorieux FH, Pettifor J, Juppner H. 2003, Nueva York, NY: Academic Press, Elsevier, 509-539.
- Collins MT: Espectro e historia natural de la displasia fibrosa del hueso. J Bone Miner Res. 2006, 21 (Suppl 2): P99-P104. 10.1359 / jbmr.06s219.
- Collins MT, Bianco P: displasia fibrosa. Primer sobre las enfermedades metabólicas óseas y los trastornos del metabolismo mineral. Editado por: Favus MJ. 2006, Washington, DC: Sociedad Americana de Investigación de Huesos y Minerales, 415-418.
- Collins MT, Shenker A: Síndrome de McCune-Albright: nuevas ideas. Curr Opin en Endocrinol y Diabetes. 1999, 6: 119 – 125. 10.1097 / 00060793-199904000-00006.
- Dorfman HD, Czerniak B: Lesiones fibroóseas. Tumores óseos. Editado por: Dorfman HD, Czerniak B. 1998, St. Louis, MO: Mosby, 441-491.
- Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, Feuillan P, Leet AI, Kushner H, Robey PG, Collins MT: Inicio, progresión y meseta de lesiones esqueléticas en la displasia fibrosa y la relación con la función Salir. J Bone Miner Res. 2007, 22: 1468-1474. 10.1359 / jbmr.070511.
- Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, Robey PG, Bianco P, Wientroub S, Collins MT: incidencia de fractura en la displasia fibrosa poliostótica y el síndrome de McCune-Albright. J Bone Miner Res. 2004, 19: 571 – 577. 10.1359 / JBMR.0301262.
- Ruggieri P, Sim FH, Bond JR, Unni KK: malignidades en la displasia fibrosa. Cáncer. 1994, 73: 1411 – 1424. 10.1002 / 1097-0142 (19940301) 73: 5 <1411 :: AID-CNCR2820730516> 3.0.CO; 2-T.
- Blanco P, Schaeverbeke T, Baillet L, Lequen L, Bannwarth B, Dehais J: Condrosarcoma en un paciente con síndrome de McCune-Albright. Informe de un caso. Rev Rhum Engl Ed. 1999, 66: 177 – 179.
- Lopez-Ben R, Pitt MJ, Jaffe KA, Siegal GP: Osteosarcoma en un paciente con síndrome de McCune-Albright y síndrome de Mazabraud. Skeletal Radiol. 1999, 28: 522 – 526. 10.1007 / s002560050556.
- Scanlon EF, Burkett FE, Sener SF, Green OC, Traisman HS, Marr TJ, Victor TA, Crist ML: Carcinoma de mama en una niña de 11 años con síndrome de Albright. Pecho. 1980
- Tanabeu Y, Nakahara S, Mitsuyama S, Ono M, Toyoshima S: Cáncer de mama en un paciente con síndrome de McCune-Albright. Cáncer de mama. 1998, 5: 175 – 178.
- Collins MT, Sarlis NJ, Merino MJ, Monroe J, Crawford SE, Krakoff JA, Guthrie LC, Bonat S, Robey PG, Shenker A: Carcinoma de tiroides en el síndrome de McCune-Albright: papel contribuyente de la activación de mutaciones Gs alfa. J Clin Endocrinol Metab. 2003, 88: 4413 – 4417. 10.1210 / jc.2002-021642.
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM: mutaciones activadoras de la proteína G estimulante en el síndrome de McCune – Albright [ver comentarios]. N Engl J Med. 1991, 325: 1688 – 1695. Schwindinger WF, Francomano CA, Levine MA: Identificación de una mutación en el gen que codifica la subunidad alfa de la proteína estimuladora G de adenilil ciclasa en el síndrome de McCune-Albright. Proc Natl Acad Sci USA. 1992, 89: 5152 – 5156. 10.1073 / pnas.89.11.5152.
- Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Hueso HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG: Mutaciones del gen GNAS1, disfunción de células estromales y cambios osteomalacicos en displasia fibrosa no de McCune-Albright. J Bone Miner Res. 2000, 15: 120-128. 10.1359 / jbmr.2000.15.1.120. Idowu BD, Al-Adnani M, O’Donnell P, Yu L, Odell E, Diss T, Gale RE, Flanagan AM: Una técnica de detección sensible a mutación sensible para mutaciones GNAS1 en casos de displasia fibrosa: el primer informe de un codón 227 mutación en el hueso. Histopatología. 2007, 50: 691-704. 10.1111 / j.1365-2559.2007.02676.x
- Riminucci M, Saggio I, Robey PG, Bianco P: displasia fibrosa como una enfermedad de células madre. J Bone Miner Res. 2006, 21 (Suppl 2): P125-131. 10.1359 / jbmr.06s224.
- Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, Brillante B, Leet AI, Riminucci M, Robey PG, Bianco P, Wientroub S, Chen CC: Un instrumento para medir la carga ósea y predecir el resultado funcional en displasia fibrosa del hueso J Bone Miner Res. 2005, 20: 219-226. 10.1359 / JBMR.041111.
- Ippolito E, Bray EW, Corsi A, De Maio F, Exner UG, Robey PG, Parrilla F, Lala R, Massobrio M, Pinggera O, Riminucci M, Snela S, Zambakidis C, Bianco P: historia natural y tratamiento de la displasia fibrosa de hueso: un estudio clinicopatológico multicéntrico promovido por la Sociedad Europea de Ortopedia Pediátrica. Revista de ortopedia pediátrica. 2003, 12: 155-177. 10.1097 / 00009957-200305000-00001.
- Kelly MH, Brillante B, Collins MT: dolor en la displasia fibrosa del hueso: cambios relacionados con la edad y la distribución anatómica de las lesiones esqueléticas. Osteoporos Int. 2007
- Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P: la histopatología de la displasia fibrosa del hueso en pacientes con mutaciones activadoras del gen Gs alfa: patrones específicos del sitio y características histológicas recurrentes. El diario de patología. 1999, 187: 249-258. 10.1002 / (SICI) 1096-9896 (199901) 187: 2 <249 :: AID-PATH222> 3.0.CO; 2-J.
- Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, Bianco P: Osteomalacic e hiperparatiroidismo cambios en la displasia fibrosa del hueso: estudios de biopsia de núcleo y correlaciones clínicas. J Bone Miner Res. 2003, 18: 1235 – 1246. 10.1359 / jbmr.2003.18.7.1235.
- Hannon TS, Noonan K, Steinmetz R, Eugster EA, Levine MA, Pescovitz OH: ¿Se pasa por alto el síndrome de McCune-Albright en sujetos con displasia fibrosa del hueso ?. J Pediatr. 2003, 142: 532 – 538. 10.1067 / mpd.2003.153. Centro de Pruebas Genéticas. [ http://www.sfh-lab.com ]
- Genome Diagnostics. [ http://www.umcutrecht.nl/subsite/genome-diagnostics/DNA-diagnostics/Gene-Tests.htm ]
- Byard RW: Consideraciones forenses en casos de neurofibromatosis: resumen. Revista de ciencias forenses. 2007, 52: 1164-1170. 10.1111 / j.1556-4029.2007.00512.x.
- Theos A, Korf BR: fisiopatología de la neurofibromatosis tipo 1. Anales de la medicina interna. 2006, 144: 842-849. Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT: visión normal a pesar del estrechamiento del canal óptico en la displasia fibrosa. N Engl J Med. 2002, 347: 1670 – 1676. 10.1056 / NEJMoa020742.
- Cutler CM, Lee JS, Butman JA, FitzGibbon EJ, Kelly MH, Brillante BA, Feuillan P, Robey PG, DuFresne CR, Collins MT: resultado a largo plazo del recubrimiento del nervio óptico y la descompresión del nervio óptico en pacientes con displasia fibrosa: factores de riesgo para la ceguera y la seguridad de la observación. Neurocirugía. 2006, 59: 1011 – 1017. discusión 1017-1018
- Feuillan PP, Shawker T, Rose SR, Jones J, Jeevanram RK, Nisula BC: Anomalías tiroideas en el síndrome de McCune-Albright: ultrasonografía y estudios hormonales. J Clin Endocrinol Metab. 1990, 71: 1596 – 1601.
- Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P: FGF-23 en la displasia fibrosa del hueso y su relación con el riñón pérdida de fosfato J Clin Invest. 2003, 112: 683 – 692.
- Terpstra L, Rauch F, Plotkin H, Travers R, Glorieux FH: mineralización ósea en displasia fibrosa poliostótica: análisis histomorfométrico. J Bone Miner Res. 2002, 17: 1949-1953. 10.1359 / jbmr.2002.17.11.1949.
- Glorieux FH, Rauch F: terapia médica de niños con displasia fibrosa. J Bone Miner Res. 2006, 21 (Suppl 2): P110-113. 10.1359 / jbmr.06s221.
- Hammami MM, Hussain SS, Vencer LJ, Butt A, al-Zahrani A: hiperparatiroidismo primario asociada a la displasia fibrosa poliostótica: ausencia de mutaciones del síndrome de McCune-Albright. J Endocrinol Invest. 1997, 20: 552 – 558.
- Barone A, Giusti A, Pioli G, Girasole G, Razzano M, Pizzonia M, Palummeri E, Bianchi G: hiperparatiroidismo secundario debido a la hipovitaminosis D afecta la respuesta de la densidad mineral ósea al alendronato en mujeres ancianas con osteoporosis: un ensayo controlado aleatorizado. Revista de la Sociedad Americana de Geriatría. 2007, 55: 752 – 757. 10.1111 / j.1532-5415.2007.01161.x.
- Giusti A, Barone A, Razzano M, Pizzonia M, Oliveri M, Palummeri E, Pioli G: Alta prevalencia de hiperparatiroidismo secundario por hipovitaminosis D en ancianos hospitalizados con y sin fractura de cadera. Revista de investigación endocrinológica. 2006, 29: 809-813.
- Reginster JY: la alta prevalencia de niveles séricos inadecuados de vitamina D y las implicaciones para la salud ósea. La investigación médica actual y la opinión. 2005, 21: 579-586. 10.1185 / 030079905X41435.
- Hashemipour S, Larijani B, Adibi H, Sedaghat M, Pajouhi M, Bastan-Hagh MH, Soltani A, Javadi E, Shafaei AR, Baradar-Jalili R, Hossein-Nezhad A: El estado de los parámetros bioquímicos en diversos grados de vitamina D deficiencia. Revista del metabolismo óseo y mineral. 2006, 24: 213-218. 10.1007 / s00774-005-0674-8.
- Stanton RP: Cirugía para la displasia fibrosa. J Bone Miner Res. 2006, 21 (Suppl 2): P105-109. 10.1359 / jbmr.06s220.
- Chapurlat RD: terapia médica en adultos con displasia fibrosa de hueso. J Bone Miner Res. 2006, 21 (Suppl 2): P114-119. 10.1359 / jbmr.06s222.
- Liens D, Delmas PD, Meunier PJ: Efectos a largo plazo del pamidronato intravenoso en la displasia fibrosa del hueso. Lanceta. 1994, 343: 953 – 954. 10.1016 / S0140-6736 (94) 90069-8.
- Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH: Efecto del tratamiento con pamidronato en niños con displasia fibrosa poliostótica del hueso. J Clin Endocrinol Metab. 2003, 88: 4569 – 4575. 10.1210 / jc.2003-030050.
- Chan B, Zacharin M: tratamiento con pamidronato de la displasia fibrosa poliostótica: fracaso para prevenir la expansión de las lesiones displásicas durante la infancia. J Pediatr Endocrinol Metab. 2006, 19: 75-80
- Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT: Displasia fibrosa en la columna vertebral: prevalencia de lesiones y asociación con escoliosis. J Bone Joint Surg Am. 2004, 86 – A (3): 531 – 537.
- Feuillan P, Calis K, Hill S, Shawker T, Robey PG, Collins MT: Tratamiento con letrozol de la pubertad precoz en niñas con el síndrome de McCune-Albright: un estudio piloto. J Clin Endocrinol Metab. 2007, 92: 2100 – 2106. 10.1210 / jc.2006-2350.
- Feuillan PP: tratamiento de la precocidad sexual en niñas con el síndrome de McCune-Albright. Precocidad sexual: etiología, diagnóstico y tratamiento. Editado por: Grave GD, Cutler GB. 1993, Nueva York: Raven Press, 243-251.
- Eugster EA, Rubin SD, Reiter EO, Plourde P, Jou HC, Pescovitz OH: tratamiento con tamoxifeno para la pubertad precoz en el síndrome de McCune-Albright: un ensayo multicéntrico. The Journal of pediatrics. 2003, 143: 60-66. 10.1016 / S0022-3476 (03) 00128-8.
- Congedo V, Celi FS: enfermedad de la tiroides en pacientes con síndrome de McCune-Albright. Pediatr Endocrinol Rev. 2007, 4 (Suppl 4): 429-433.
- Akintoye SO, Kelly MH, Brillante B, Cherman N, Turner S, Butman JA, Robey PG, Collins MT: Pegvisomant para el tratamiento del exceso de hormona de crecimiento mediada por gsp en pacientes con síndrome de McCune-Albright. J Clin Endocrinol Metab. 2006, 91: 2960-2966. 10.1210 / jc.2005-2661.
- Chanson P, Salenave S, Orcel P: Síndrome de McCune-Albright en la edad adulta. Pediatr Endocrinol Rev. 2007, 4 (Suppl 4): 453-462.
- Galland F, Kamenicky P, Affres H, Reznik Y, Pontvert D, Le Bouc Y, Young J, Chanson P: síndrome de McCune-Albright y acromegalia: efectos de la radioterapia hipotalámico-hipofisaria y / o pegvisomant en pacientes con resistencia al análogo de la somatostatina. J Clin Endocrinol Metab. 2006, 91: 4957 – 4961. 10.1210 / jc.2006-05
- Ehrig U, Wilson DR: Displasia fibrosa ósea e hiperparatiroidismo primario. Ann Intern Med. 1972, 77: 234 – 238.
- Leslie WD, Reinhold C, Rosenthall L, Tau C, Glorieux FH: displasia fibrosa panostótica. Una nueva displasia craneotubular. Medicina nuclear clínica 1992, 17: 556 – 560. 10.1097 / 00003072-199207000-00005.
- Feuillan PP: Síndrome de McCune-Albright. Curr Ther Endocrinol Metab. 1997, 6: 235 239.
- Defilippi C, Chiappetta D, Marzari D, Mussa A, Lala R: diagnóstico de imagen en el síndrome de McCune-Albright. J Pediatr Endocrinol Metab. 2006, 19 (Suppl 2): 561-570.
- Silva ES, Lumbroso S, Medina M, Gillerot Y, Sultán C, Sokal EM: Demostración de mutaciones de McCune-Albright en el hígado de niños con alta colestasis progresiva gammaGT. Revista de hepatología. 2000, 32: 154 – 158. 10.1016 / S0168-8278 (00) 80202-0.
- Zimmerman D: hipertiroidismo fetal y neonatal. Tiroides. 1999, 9: 727 – 733.
- Schwartz RA, Spicer MS, Leevy CB, Ticker JB, Lambert WC: Displasia fibrosa cutánea: una forma incompleta del síndrome de McCune-Albright. Dermatología (Basilea, Suiza). 1996, 192: 258 – 261.
- Pierini AM, Ortonne JP, Floret D: [Manifestaciones cutáneas del síndrome de McCune-Albright: informe de un caso (transl del autor)]. Annales de dermatologie et de venereologie. 1981, 108: 969 – 976.
- Grumbach MM, Styne DM. Puberty: Ontogeny, Neuroendocrinology, physiology and disorders. En: Wilson JD, Foster DW, Kronenberg HM, Larsen PR. Willians Textbook of Endocrinology. 9th ed. Philadelphia: Saunders; 1998.Págs.1509-625.
- McCune DJ. Osteitis fibrosa cystica: the case of a nine-year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child.1936; 52:743-4.
- Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disseminata, areas of pigmentation, and endocrine dysfunction, with precocious puberty in females: report of 5 cases. N Engl J Med. 1937; 216(17):727-46.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis. 2008; 3:12. 5. Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012; 7(Suppl 1):S4.
- Tufano M, Ciofi D, Amendolea A, Stagi S. Auxological and Endocrinological Features in Children with McCune Albright Syndrome: A Review. Front Endocrinol (Lausanne). 2020; 11:522. Congedo V, Celi FS. Thyroid disease in patients with McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007; 4(Suppl 4):429-33.
- Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014; 99(6):1955-69.
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001; 22(5):675-705. Lumbroso S, Paris F, Sultan C, European Collaborative Study. Activating Gs Mutations: Analysis of 113 Patients with Signs of McCune-Albright Syndrome. J Clin Endocrinol Metab. 2004; 89(5):2107-13.
- Brito V, Latronico A, Arnhold I, Mendonça BB. Update on the Etiology, diagnosis and therapeutic management of sexual precocity. Arq Bras Endocrinol Metab. 2008; 52(1):1831. 12. Zacharin M. The Spectrum of McCune-Albright syndrome. Ped Endocrinol Rev. 2007; 4(Suppl 4):412-8. 13.
- Neyman A, Eugster EA. Treatment of Girls and Boys with McCune-Albright Syndrome with Precocious Puberty – Update 2017. Pediatr Endocrinol Rev. 2017; 15(2):136-41. 14.
- Corica D, Aversa T, Pepe G, De Luca F, Wasniewska M. Peculiarities of Precocious Puberty in Boys and Girls wth McCune-Albright Syndrome. Front Endocrinol (Lausanne). 2018; 9:337. Escobar ME, Rivarola MA, Bergadá C. Plasma Concentration of estradiol-17β in premature thelarche and in different types of sexual precocity. Acta Endocrinol (Copenh). 1976; 81(2):351-61. Ballerini MG, Ropelato MG, Gruñeiro LO, Bozzo G, et al. Estradiol (E2): determinación de la sensibilidad funcional de inmunoensayos radioisotópicos (IEs) y no radioisotópicos. Rev Argent Endocrinol Metab. 1999; 36(Suppl):86.
- Sizonenko PC, Burr IM, Kaplan SL, Grumbach MM. Hormonal Changes in Puberty II. Correlation of serum luteinizing hormone and follicle stimulating hormone with stages of puberty and bone age in normal girls. Pediatr Res. 1970; 4(1):36-45.
- Ropelato MG, Escobar ME, Gottlieb S, Bergadá C. Gonadotropin secretion in prepubertal normal and agonadal children evaluated by ultrasensitive time-resolved immunofluorometric assays. Horm Res. 1997; 48(4):164-72.
- Domené HM, Sansó G, Gryngarten M, Escobar ME. AlleleSpecific Amplification (AS-PCR) for Point Mutation Screening of G Protein α-Subunit (Gsα) in McCune-Albright Syndrome (MAS). Pediatr Res. 1999; 45:441.
- Lejarraga H, Orfila G. Estándares de peso y estatura para niñas y niños argentinos desde el nacimiento hasta la madurez. Arch Argent Pediatr. 1987; 85(4):209-22.
- Collins MT. Spectrum and natural history of fibrous dysplasia of bone. J Bone Miner Res. 2006, 21(Suppl 2):P99104.
- Sousa G de, Wunsch R, Andler W. Precocious pseudopuberty due to autonomous ovarian cysts: A report of ten cases and long-term follow-up. Hormones (Athens). 2008; 7(2):170-4.
- Murad F, Haynes RC. Estrógenos y Progestágenos. En: Goodman Gilman A, Goodman LS, Rall TW, Murad F. Goodman y Gilman: Las Bases farmacológicas de la Terapéutica. 7ª ed. Buenos Aires; Panamericana; 1986. Págs.1341-65.
- Eugster EA, Rubin SD, Reiter EO, Plourde P, et al. Tamoxifen treatment for precocious puberty in McCune-Albright syndrome: a multicenter trial. J Pediatr. 2003; 143(1):60-6.
- Passone CB, Kuperman H, Menezes-Filho HC, Esteves LSO, et al. Tamoxifen Improves Final Height Prediction in Girls with McCune-Albright Syndrome: A Long FollowUp. Horm Res Paediatr. 2015; 84(3):184-9.
- Feuillan P, Calis K, Hill S, Shawker T, et al. Letrozole treatment of precocious puberty in girls with the McCuneAlbright syndrome: a pilot study. J Clin Endocrinol Metab. 2007; 92(6):2100-6.
- Estrada A, Boyce A, Brillante B, Guthrie L, et al. Long-term outcomes of letrozole treatment for precocious puberty in girls with McCune-Albright syndrome. Eur J Endocrinol. 2016; 175(5):477-83.
- Gesmundo R, Guanà R, Valfrè L, De Sanctis L, et al. Laparoscopic management of ovarian cysts in peripheral precocious puberty of McCune-Albright syndrome. J Pediatr Endocrinol Metab. 2006; 19(Suppl 2):571-5.
- Escobar ME, Gryngarten MG, Domenè H, Ropelato MG, et al. Persistence of autonomous ovarian activity after discontinuation of therapy for precocious puberty in McCune-Albright syndrome. J Pediatr Adolesc Gynecol. 1997; 10(3):147-51.
- Lala R, Andreo M, Pucci A, Matarazzo P. Persistent hyperestrogenism after precocious puberty in young females with McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007; 4(Suppl 4):423-8.
- Sotomayor K, Iñiguez G, Ugarte F, Villarroel C, et al. Ovarian function in adolescents with McCune-Albright syndrome. J Pediatr Endocrinol Metab. 2011; 24(7-8):525-8.
- Riminucci M, Robey PG, Bianco P. The pathology of fibrous dysplasia and the McCune-Albright syndrome. Pediatr Endocrinol Rev. 2007; 4(Suppl 4):401-11.
- Boyce AM, Collins MT. Fibrous Dysplasia/McCuneAlbright syndrome: a rare, mosaic disease of Gα s activation. Endocr Rev. 2020; 41(2):345-70.
- Majoor BC, Appelman-Dijkstra NM, Fiocco M, van de Sande MA, et al. Outcome of long-term bisphosphonate therapy in McCune-Albright syndrome and polyostotic fibrous dysplasia. J Bone Miner Res. 2017; 32(2):264-76.
- Lala R, Matarazzo P, Andreo M, Marzari D, et al. Bisphosphonate treatment of bone fibrous dysplasia in McCune-Albright syndrome. J Pediatr Endocrinol Metab. 2006; 19(Suppl 2):583-93. 36. Shah KN. The diagnostic and clinical significance of caféau-lait macules. Pediatr Clin North Am. 2010; 57(5):1131-53.