INFORMACIÓN BÁSICA.- Displasia Diastrófica (OMIM: 222600/CIE-10:Q77.5/ORPHA:628).- La displasia diastrófica (DD) también llamada enanismo diastrófico es una enfermedad rara, de origen genético, caracterizada por estatura baja con extremidades cortas (la estatura final de un adulto es de 120cm +/- 10cm), y malformaciones de las articulaciones, que conducen a contracturas articulares múltiples (afectando principalmente a hombros, codos, articulaciones interfalángicas y caderas).
Frecuencia.- La prevalencia muy rara se estima en 1/100.000. La enfermedad afecta tanto a hombres como a mujeres.
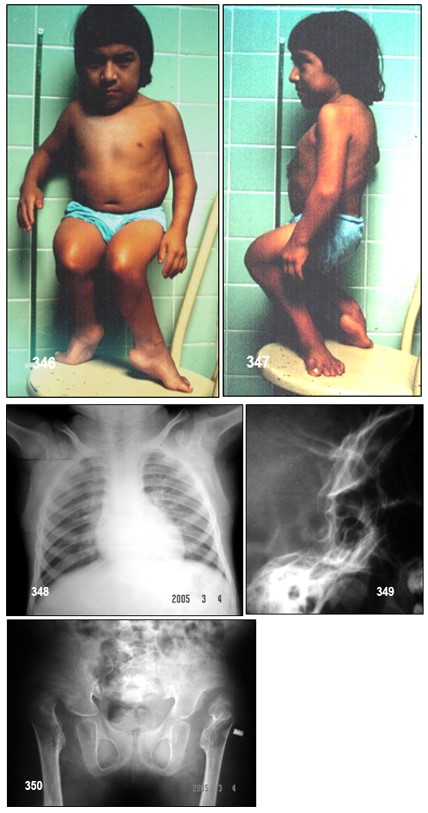
Manifestaciones clínicas.- En el nacimiento, los niños presentan pie zambo bilateral (pie bot), extremidades cortas, deformación de las muñecas y pulgares en aducción. Fisura palatina y mandíbulas hipoplásicas son también características habituales. En los primeros meses de vida aparecen quistes en el oído externo. Se manifiesta por estatura baja, extremidades cortas (severa rizoacromelia) y malformaciones en diferentes articulaciones, entre ellas hombros, codos, interfalángicas (mano “haciendo dedo”) y caderas. Los niños afectados presentan crecimiento lento y frecuentemente escoliosis de desarrollo progresivo. El crecimiento es lento y la escoliosis es frecuente y se desarrolla de manera progresiva. Las deformaciones de las articulaciones son graves y pueden conducir a una limitación del movimiento de la articulación, o bien a una hiperlaxitud. La enfermedad puede manifestarse con gravedad variable, dependiendo de los casos, existen formas graves como la ausencia de radio (1/30.000 nacidos vivos) y otras con síntomas más leves que se suele diagnosticar más tardíamente.
La DD puede asociarse a otras entidades como Síndrome de Holt-Oram, el síndrome de trombocitopenia-radio ausente.
Herencia.- El síndrome se transmite de forma autosómica recesiva (riesgo relativo: 25%) y está provocado por mutaciones en el gen SLC26A2, 5q31-q34 (o transportador de sulfato de la displasia diastrófica; DTDST), que codifica para un transportador de sulfato con expresión predominante en el cartílago. Mutaciones en el mismo gen están implicadas en una forma moderada de displasia epifisaria y en numerosas afecciones letales, como la acondrogénesis de tipo 1b y la atelosteogénesis de tipo 2 (ver estos términos).
Etiología.- La enfermedad está causada por una mutación en el gen SLC26A2, también llamado DTDST, situado en el cromosoma 5 (5q32-q33.1). Este gen codifica una proteína esencial para el desarrollo adecuado del cartílago y su conversión a hueso.
La proteína codificada por el gen SLC26A2, es una proteína transmembrana relacionada con el transporte de sulfato, su alteración está implicada también en otras enfermedades relacionadas, entre ellas la acondrogénesis 1B y la displasia epifisaria múltiple recesiva. El fallo en esta proteína transportadora provoca una reducción del transporte de sulfato en las células del cartílago conocidas como condrocitos, lo que ocasiona baja sulfatación de los proteoglicanos, ocasionando la formación anómala de los cartílagosy en consecuencia la talla baja y las deformidades óseas.
Se han identificado más de 20 mutaciones en el gen SLC26A2 en las personas portadoras de DD.
Diagnóstico.- El diagnóstico es radiológico: huesos tubulares cortos y macizos, metáfisis anchas, aspecto ovoide y corto del primer metacarpiano, subluxación del pulgar (pulgar del autoestopista) y subluxación de las vértebras cervicales. El diagnóstico prenatal se puede sospechar en base a los hallazgos en la ecografía (pie zambo y extremidades cortas). Para filiar la mutación se debe hacer estudios para detección de las mutaciones asociadas a la DD, mediante la amplificación completa por PCR de los exones del gen DTDST (SLC26A2), y su posterior secuenciación.
El manejo debe incluir un seguimiento cuidadoso y eventualmente una corrección de la escoliosis progresiva y una corrección quirúrgica de las malformaciones articulares. En ausencia de complicaciones graves (comprensión de la médula espinal) asociadas a malformaciones de la columna vertebral, la esperanza de vida es buena pero la estatura corta y las malformaciones son habitualmente graves.
Bibliografía
- Federación Española de Enfermedades Raras: Displasia diastrófica.
- Bueno M, Bueno-Lozano M, Bueno AL. Osteocondrodisplasias. En: Pombo M (ed). Tratado de Endocrinología Pediátrica, 2ª ed. Madrid:
- Díaz de Santos, 1997; págs. 331-348.
- OMIM: Diastrophic dysplasia.
- Orphanet: Enanismo diastrófico.
- http://www.orpha.net/consor/cgibin/Disease_Search.php?lng=ES&data_id=209&Disease_Disease_Search_diseaseType=ORPHA&Disease_
- Disease_Search_diseaseGroup=628&Malattia(e)/%20gruppo% 20di%20malattie=Displasia-diastrofica&title=Displasia-diastrofica&search=Disease_Search_Simple