INFORMACIÓN BÁSICA.- Displasia Epifisaria Múltiple (OMIM: 251/cie-10:Q77.3/OMIM:607131).- La Displasia Epifisaria Múltiple (DEM) es una enfermedad genética que afecta el crecimiento y remodelación del hueso que forma parte del grupo de las displasias óseas. Se trata de una anomalía en el desarrollo de las epífisis del hueso, en la que hay una falta de osificación del extremo creciente del hueso. En consecuencia se producen deformidades óseas que afectan a las articulaciones y producen artrosis tempranas. Se transmite de forma autosómica dominante.
Clasificación.- Las DEMs se caracterizan por anomalías epifisarias responsables de dolores articulares de aparición temprana, osteocondrosis repetidas y artrosis temprana. Las DEMs son un grupo heterogéneo de enfermedades con expresión variable y se clasifican en 6 tipos. La EDM1 es la forma mejor caracterizada clínicamente y se asocia a problemas de la marcha y dolores de aparición temprana, además de estatura baja moderada. La principal complicación es la artrosis temprana de cadera.
Existe una forma precoz de DEM o del recién nacido llamada enfermedad de Conradi, siendo incompatible con la vida. Se caracteriza por talla corta, retraso mental, cataratas congénitas y calcificaciones traqueales.
La forma de DEM del adulto, también conocida como enfermedad de Fairbank. Existe también una forma más leve conocida como Enfermedad de Ribbing
Prevalencia.- La prevalencia media se estima alrededor de 1/20.000.
Genética.- La EDM1 se transmite de forma autosómica dominante y está causada por mutaciones en el gen COMP; 19p13.1, que codifica para la proteína oligomérica de la matriz del cartílago. Las otras formas de EDM con herencia dominante, no están tan bien caracterizadas clínicamente, pero en algunos casos se han identificado mutaciones responsables de la enfermedad en genes que codifican para diversos componentes de la matriz extracelular del cartílago: COL9A2 (1p33-p32.2) para EDM2, COL9A3 (20q13.3) para EDM3 y COL9A1 (6q13) para EDM6 (EDMs debido a anomalías en el colágeno tipo 9, y MATN3 (2p24-p23) para EDM5. Se ha descrito una forma atípica de EDM (EDM4) caracterizada por pie zambo y rótula «en doble capa»; su transmisión es de modo autosómico recesivo y está provocada por mutaciones en el gen SLC26A2 (5q32-q33.1).
Casi la mitad de todas las personas con DEM no tendrán un cambio identificable en ninguno de estos genes conocidos. Es por esta razón, en particular, que la DEM sigue siendo un diagnóstico clínico y radiológico.
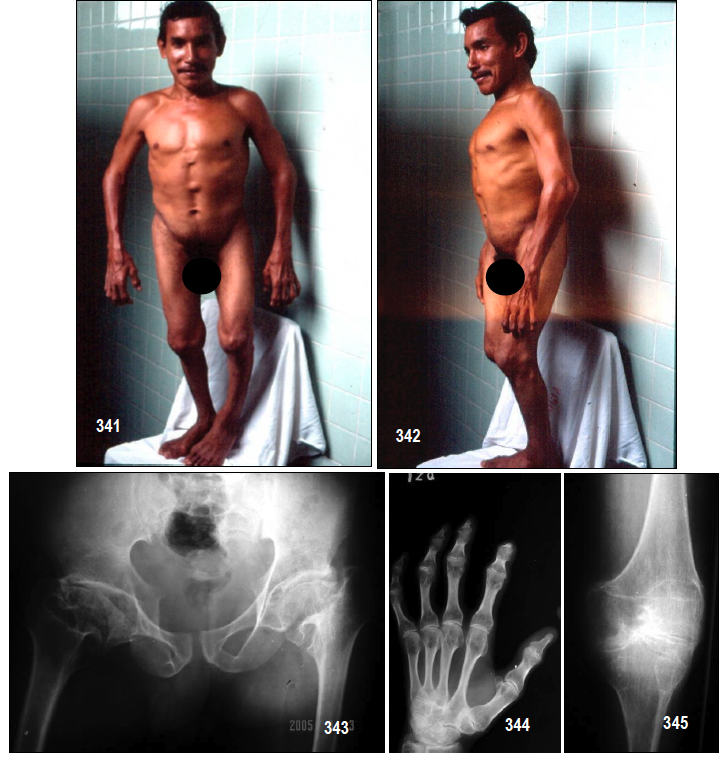
Características clínicas.- En la DEM del adulto, a pesar de estar presente la enfermedad ya en el nacimiento, las manifestaciones no suelen aparecer hasta el 1º-10º año de vida. Se trata de sujetos cortos de estatura y con inteligencia normal. Afecta, donde las deformidades epifisarias dan lugar a disfunción articular en forma de rigideces y/o limitaciones articulares, y posteriormente artrosis, muy frecuentemente en caderas, lo que les lleva a precisar con frecuencia una prótesis de cadera en la edad adulta temprana.
Los pacientes se ven afectados de fuertes dolores articulares que van aumentando en intensidad con el tiempo y extendiéndose limitando gravemente su movilidad y bienestar.
Algunas formas de displasia se limitan principalmente a la epífisis femoral (displasia de Meyer). Además, también se han descrito otros síndromes caracterizados por la asociación de DEM con otras manifestaciones clínicas, como miopía, compromiso del vítreo, sordera y dismorfia facial. El diagnóstico se basa en la identificación de los signos radiológicos.
Un rasgo clínico típico que permite distinguirla de la enfermedad de Morquio, (una mucopolisacaridosis que también presenta talla corta y deformidades articulares) es que en la DEM la columna vertebral no suele verse afectada. En el Morquio la afectación vertebral es frecuente y típica. La DEM se asocia a otras enfermedades congénitas
Diagnóstico.- A diferencia de muchas displasias óseas, la manifestación de la displasia epifisiaria múltiple podría retrasarse y frecuentemente el diagnóstico no se hace sino hasta que el niño tiene entre 2 a 10 años de edad. El diagnóstico a menudo puede retardarse incluso más. Los primeros indicadores de la presencia de este trastorno son la marcha miopática (de pato), dificultades para correr, cojeo, rigidez de articulaciones, dolor articular (particularmente de la cadera y/o rodillas), fatiga precipitada por las actividades, y/o una desaceleración sutil en la velocidad de crecimiento.
Las alteraciones radiológicas suelen ser la clave del diagnóstico. Los centros de osificación epifisarios suelen aparecer tarde, (retraso de edad ósea), y aparecen con una forma irregular, o incluso fragmentados, en las formas más graves.
Muy importante comprobar que la afectación es múltiple.
- -Caderas: La epífisis proximal del fémur se deforma, aplanándose, con parecido a una coxa vara. Aparece artrosis precoz, aunque sin quistes periarticulares.
- -Rodillas: Cuadrangulación de los cóndilos del fémur. También es frecuente el genu valgo, (pudiendo ser planteable una osteotomía correctora).
- -Tobillos: Un signo típico radiológico de esta enfermedad es la oblicuidad del pilón de la tibia.
- -Manos y pies: Los metacarpianos y metatarsianos son cortos y con un crecimiento atrasado.
- -Codos, hombros y mandíbula.
La afectación de columna vertebral es menos intensa que en el Morquio, pero no imposible, pudiendo aparecer muescas e irregularidades en las apófisis anulares de T12 y L1.
Existen diferencias radiológicas sutiles entre las causas moleculares conocidas de la DEM. Por ejemplo, aquellos con mutaciones del COMP tienen más compromiso de cadera, aquellos con mutaciones del col9 tienen más anomalías a nivel de la rodilla y aquellos con mutaciones del DTDST tienen una doble rótula característica. Estas y otras diferencias pueden ser utilizadas para orientar la evaluación diagnóstica molecular. Lo que es más importante, si bien las características de las diferentes formas de DEM coinciden, ciertas distinciones son evidentes en base al gen que origina el trastorno:
- Aquellas personas con una anomalía en la COMP suelen ser de estatura más baja, mientras que aquellos en los que el trastorno es causado por MATN3 son asociados con una estatura normal.
- Las mutaciones del Col9 suelen resultar en un inicio de las manifestaciones articulares más leve y tardío que otras formas.
- La miopatía pareciera estar asociada con la displasia epifisiaria múltiple causada por cambios en MATN3 o col9A3.
- La rigidez articular, y no tanto hipermovilidad de articulaciones, se manifiesta cuando la displasia epifisiaria múltiple es causada por mutaciones en DTDST.
- El pie equinovaro es común cuando se trata de displasia epifisiaria múltiple causada por mutaciones en DTDST pero casi nunca se observa en las otras formas
Tratamiento.- Al no existir cura para la DEM el tratamiento habitual incluye principalmente fisioterapia y manejo ortopédico, aunque el médico puede recomendar tratamiento quirúrgico, ya sea para corregir las deformidades, (osteotomías femorales distales) o para implantar prótesis, (cadera). La DEM es una de las entidades que se encuentran actualmente en evaluación para definir la utilidad de la hormona de crecimiento humana biosintética y que requiere para su uso de un estudio prospectivo de seguridad y eficacia.
Otro tipo de Displasia Epifisaria Múltiple Tipo AlGgazali.- La displasia epifisaria múltiple tipo Al-Gazali es una displasia esquelética caracterizada por una displasia epifisaria múltiple (consulte este término), macrocefalia y dismorfismo facial.
Epidemiología.- Ha sido descrita en 4 niños de una familia de Omán.
Descripción clínica.- Los rasgos dismórficos consisten en macrocefalias con abombamiento frontal, hipertelorismo, región malar plana, orejas de implantación baja y cuello corto.
Etiología.- El gen de la enfermedad ha sido localizado en la región telomérica del brazo largo del cromosoma 15 y la enfermedad se transmite de forma autosómica recesiva.
Bibliografía
- Bueno M, Bueno-Lozano M, Bueno AL. Osteocondrodisplasias. En: Pombo M (ed). Tratado de Endocrinología Pediátrica, 2ª ed. Madrid: Díaz de Santos, 1997; págs. 331-348.
- Trehan R, Dabbas N, Allwood D, Agarwal M, Kinmont C (2008). «Arthroscopic decompression and notchplasty for long-standing anterior cruciate ligament impingement in a patient with multiple epiphyseal dysplasia: a case report». J Med Case Reports 2: 172.
- Ikegawa S. Genetic analysis of skeletal dysplasia: recent advances and perspectives in the post-genome-sequence era. J Hum Genet 2006; 51:581-586.
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=251
- Spranger J, Winterpacht A, Zabel B. The type II colagenpathies: A spectrum of chondrodysplasias. Eur J Pediatr 1994; 153:56-65.
- Nishimura G, Fukushima Y, Aihara T, Ohashi H, Nishimoto H, Nishimura J. Previously undescribed spondyloepiphyseal dysplasia associated with craniosynostosis, cataracts, cleft palate and mental retardation. Amer J Med Gen 1998; 77:1-7
- Hall C. Spondyloepimetaphyseal dysplasia with multiple dislocations (Hall type): three further cases and evidence of autosomal dominant inheritance. J Med Genet 2002; 39:666-670
- Vikkula M, Marinam E, Zhidkova N, Tiller G, Golding M, Van Beersum S, De Waal M, Van der Hoogen F, Ropers H, Mayne R, Cheah K, Olsen B, Warman M, Brunner H. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995; 80:431-437.