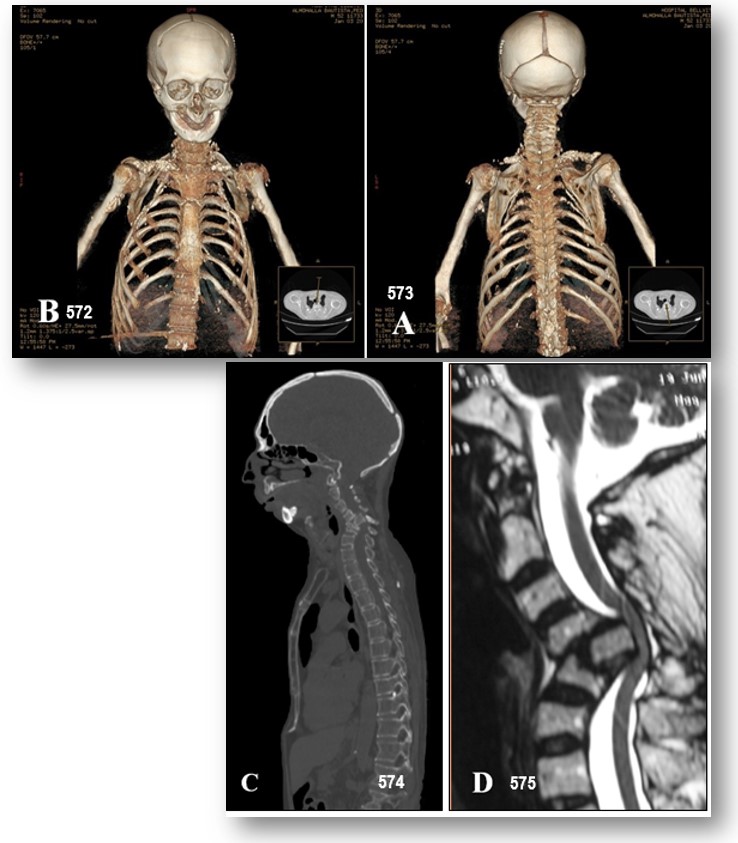
Imágenes 572-573.- Síndrome Hanhart.- Las radiografías cervicales revelaron espondiloptosis en la vértebra C5 con colapso asociado. Imágenes 574-575 .- Síndrome Hanhart.- Los estudios de neuroimagen (TC y RM) confirmaron la espondiloptosis con severa compresión medular cervical severa y estenosis de canal y un amplio espectro de anomalías del desarrollo del esqueleto con hipoplasia de clavícula, escápulas y esternón (Imagen 574). (Gentileza de los Drs. Carlos Valencia Calderón, Ana Calderón Valdiviezo, Catalina Vásquez Hahn, Ecuador)
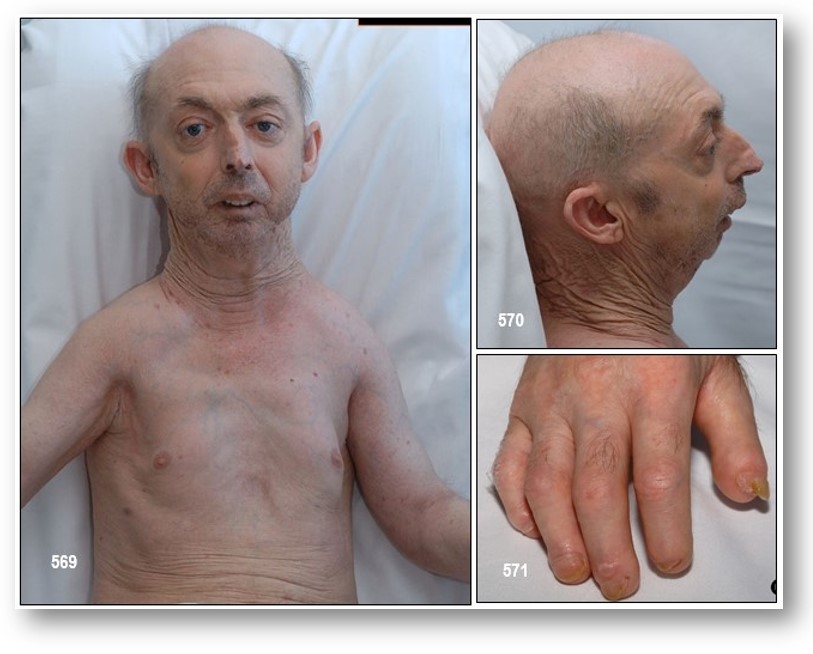
Comentarios de los Autores: El paciente descrito aquí presentó un síndrome de Hanhart con tetraparesia secundaria a espondiloptosis de la quinta vértebra cervical en el que no se indicó tratamiento quirúrgico. El síndrome de Hanhart es un raro defecto del nacimiento en el que las señales más obvias son una la lengua incompletamente desarrollada (hipoglosia); hipoplasia de dedos de manos y pies (hipodactilia); brazos y/o piernas malformados (peromelia); y una mandíbula sumamente pequeña (micrognatia). La severidad de estas anormalidades físicas varía grandemente de un caso a otro. La causa de síndrome de Hanhart es desconocida. Casos de hipogenesis de miembro y afectación oromandibular (síndrome de Hanhart) han sido reportados (1-4), sin embargo, y a nuestro mejor conocimiento, éste es el primer caso informado asociado a las anomalías espinales. Sin embargo, nosotros pensamos que este síndrome no es genéticamente o históricamente asociado con las anormalidades espinales y que este paciente pudo haberse caído hace años y pudo haber tenido una lesión cervical no diagnosticada y que ha progresado con el tiempo. El síndrome de Hanhart es extraordinariamente infrecuente, pero es importante describirlo para que sea posible detectarlo y de esta manera contribuir al esclarecimiento de sus posibles factores etiológicos, hereditarios o ambientales. Debe buscarse los defectos orales al examinar a un paciente con malformaciones distales de las extremidades.
INFORMACION BÁSICA: Síndrome de aglosia-adactilia o Síndrome de Hanhart (SH) (OMIM 103300/ ORPHA:989).– Es una enfermedad extremadamente rara caracterizada por hipoplasia de la lengua y de los miembros (peromelia), de etiología posiblemente genética, cuyo diagnostico es clínico-radiológico y cuyo tratamiento es multidisciplinario. La patogenia del daño tisular no es muy clara. Los altos niveles de plasmáticos de tirosina y metabolitos (ácidos fenólicos) se han relacionado, pero esta asociación no ha sido confirmada aún.
Las malformaciones más destacadas son orofaciales y adactilia. Otros hallazgos ocasionales son un peso bajo al nacer y parálisis de los nervios craneales. No suele presentarse retraso mental. La comprensión del desarrollo embriológico es importante para entender la asociación de ciertos rasgos en este síndrome.
El primer caso de aglosia (ausencia de lengua) y anomalías de miembros asociadas se describió en 1932, y en 1950 Hahnart describió 3 casos de la misma asociación, dando su nombre al síndrome. Varios casos similares se han publicado desde entonces, permitiendo una descripción mejor de la asociación, que incluye varias combinaciones de anomalías craneofaciales, de miembros, y otras menos frecuentes.
Sinónimos: Hipoglosia Hipodactilia, Aglosia-Adactilia, Síndrome de Peromelia con Micrognatia y microstomia.
Prevalencia.- La prevalencia estimada es de 1 en 500.000.
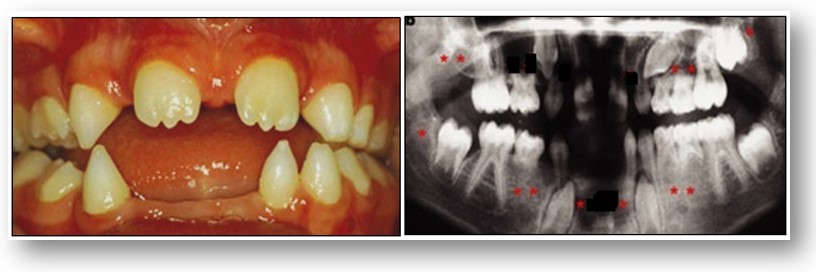
Manifestaciones clínicas: Después de una infancia normal, se evidencia un retraso mental entre los 1.5 años y 6 años, con un retraso en el crecimiento. Se observan unos rasgos faciales típicos y ocasionalmente braquicefalia. Depósitos de lípidos en los pechos y en la región abdominal, con un retraso en el crecimento de las gónadas y características sexuales secundarias. Las anomalías craneofaciales incluyen microstomia, micrognatia, hipoglosia, fisuras o mamelones aberrantes en la lengua, hipodoncia mandibular, fisura palatina, parálisis de nervios craneales incluyendo secuencia de Möebius, nariz ancha, telecanto, defectos en los párpados inferiores, y asimetría facial. Los defectos de los miembros están representados por hipoplasia de varios grados, desde la ausencia de las falanges distales hasta la adactilia total o amputación parcial de los miembros, con o sin sindactilia. Generalmente están implicados los cuatro miembros.
– Otras malformaciones. Aunque menos comunes son la gastrosquisis y fusión esplenogonadal. Habitualmente la inteligencia y la estatura son normales, pero se han descrito casos de retraso mental. Algunos de los síntomas listados pueden confundirse con el síndrome de Nager, o disostosis acrofacial. Ambos síndromes se diferencian por el tipo de dismorfia facial (los pabellones auriculares en el síndrome de Nager están malformados y la hipoplasia malar está asociada a fisuras palpebrales de inclinación descendente) y por el tipo de anomalías de los miembros, que son preaxiales a menudo en el Nager y reducciones transversas en la aglosia-adactilia.
La severidad de estas anomalías físicas descritas varía mucho, según los casos. Los niños con esta enfermedad tienen a menudo algunos de los síntomas, pero no todos. Se desconoce la causa del síndrome de Hanhart.
Genética del síndrome: Entre 1932 y 1991, se han descrito en la literatura cerca de 30 casos de síndrome de Hanhart, y algunos casos de recurrencia intrafamiliar han llevado a la hipótesis de una mutación recesiva en un gen que se encuentra en el cromosoma 16q22.1-22.3 q. Según se documentaron 100 casos, se observó que la mayoría de los cuales en los italianos. Aunque la etiologia de este síndrome poco frecuente sigue siendo desconocida y del medio ambiente y los factores genéticos son los estudios. Si embargo, esta hipótesis genética, debido a una deficiencia de la enzima tirosina aminotransferasa, ya negada en 1973, se ha sustituido ahora por una hipótesis más plausible: las anomalías probablemente son la consecuencia disruptiva de lesiones hemorrágicas durante el desarrollo, y el presunto problema vascular parece ocurrir más en las regiones distales, como en los miembros distales, lengua, y ocasionalmente en partes del cerebro.
La recogida de muestras de biopsia de vellosidades coriónicas (BVC), cuando se lleva a cabo antes de la semana 10 de amenorrea, se ha asociado a este trastorno, lo que proporciona mayor evidencia de la hipótesis disruptiva vascular. Generalmente, los rasgos descritos después de la BVC son de menor severidad que aquellos descritos en el síndrome de Hanhart, pero probablemente pertenezcan al mismo espectro de anomalías.
Ya que esta asociación entre la práctica de BVC precoz y la disgenesia de miembros oromandibular está confirmada, los BVCs se realizan más tarde durante el embarazo, y se describen muy raramente en la literatura casos de esta malformación disruptiva. Pueden aparecer dificultades del habla y de la alimentación a edades tempranas. Se puede precisar cirugía plástica y ortopédica para las anomalías de miembros.
En el síndrome de Hanhart, la gravedad varía con el significado de las diferentes alteraciones extremidades, de peromelia completas a la ausencia distal de un dedo, sindactilia o hipoplasia ungueal. Micrognatia y microglosia pueden ser graves y se asocia con fisura palatina, sinequias, singnactilia y oligodoncia (véase las imágenes con fines didácticos). Se puede producir la parálisis congénita de nervios craneales unilaterales o bilaterales.
La capacidad intelectual se mantiene por lo general.
Por otro lado, hay denuncias de mutaciones de novo, otros de causa desconocida, y en otros casos asociados a consanguinidad paterna.
Diagnóstico diferencial.- El diferencial de este síndrome deben considerarse la ectrodactilia y las parálisis aisladas de nervios craneales, también el síndrome de Poland Mobius, que es la combinación completa o incompleta de sinbraquidactilia, defecto torácico y paralisis de un nervio craneal. El síndrome de bandas amnióticas se distingue de la agenesia primaria de extremidades por la presencia de cicatrices activas en el recién nacido, con anillos que constrifien e interruption de los dermatoglifos por la amputación.
Prevención: Hacer hincapié en la imposibilidad de la prevención, y los pacientes pueden morir a causa de la aspiración de alimentos, los padres de los niños que son un subsidiario de apoyo psicológico. Las posibilidades de tratamiento logopedical y prótesis de extremidad peromélica deben ser evaluados.
Bibliografía
- Hanhart syndrome Genetic and Rare Diseases Information Center (GARD)
- Castillo ST, Rojas JZ, Monasterio de Los Ángeles. Síndrome de Hanhart. Rev Chil Pediatr. 1985, 56 (3):180-183.
- Dra. Silvia Castillo T.; Dr. Jorge Rojas Z.; Dr. Luis Monasterio A. Síndrome de Hanhart. . Rev. Chil. Pediatr. 56(3): 180 183, 1985.
- Paige DG, Clayton P, Bowron A, Harper JI. Síndrome de Richner-Hanhart (oculo cutaneoustyrosinaemia, tirosinemia tipo II). Revista de la Sociedad Real de Medicina. 1992, 85:759-760.
- Grompe M, St Louis M, Demers S, Al Dhalimy M, Leclerc B, Tanguay R: A Single Mutation of the Flumarylacetoacetate Hydrolase Gene in French Canadians with Hereditary Tyrosinemia Type I. NEJM 1994; 331: 353-7.
- Croffie J, Gupta S, Chong S, Fitzgerald J: Tyrosinemia Type I should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatr 1999; 103: 675-8.
- Bergeron A, D’Astous M, Timm D, Tanguay R: Structural and Functional Analysis of Missense Mutations in Flumarylacatoacetate Hydrolase, the Gene Deficient in Hereditary Tyrosinemia Type I. J Biol Chem 2001; 276: 15225-