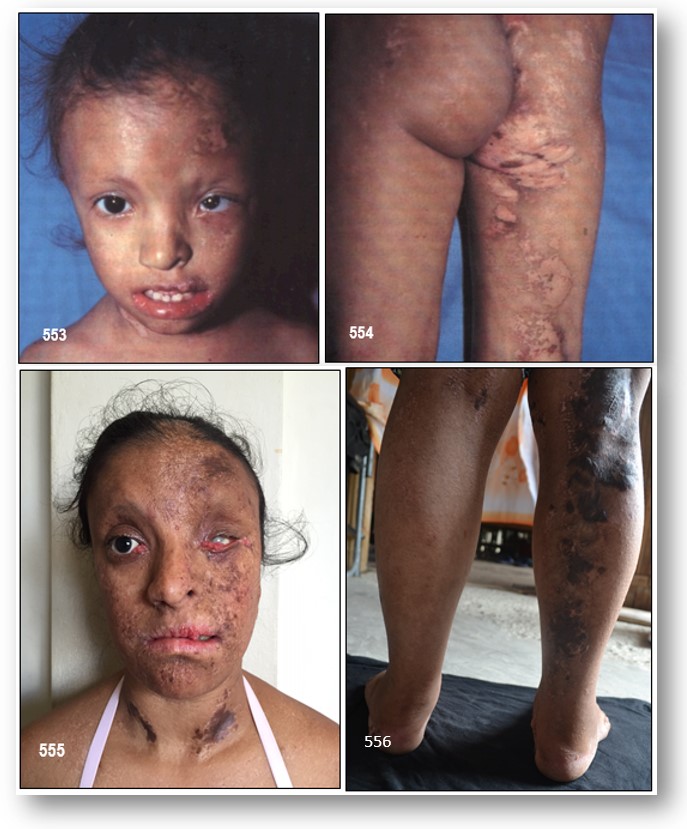
INFORMACIÓN BÁSICA.- Síndrome de Goltz o Hipoplasia Focal Dérmica (OMIM: 305600/ ORPHA:2092/CIE-10:Q82.8) (HFD).- Gotz y cols. en 1962 y Gorlin y cols. en 1963 definen este síndrome que consiste en una asimetría facial, del tronco y extremidades; atrofia, telangiectasias e hiperpigmentación de las líneas de la piel; depósitos de grasa de manera superficial en la piel acompañándose además de papilomas múltiples en muco-cutaneos, anormalidades del esqueleto, especialmente de las extremidades. El primer caso de esta enfermedad fue reportada por Jassner en 1921. Una excelente descripción de algunos casos fueron denunciados por Braun-Falco y Hofmann. Ferrara y col lo reportaron en 1967, inicialmente como síndrome de Bloom siendo posteriormente reclasificado.
El síndrome de HFD es una rara enfermedad congénita multisistémica que, como ocurre a menudo con otros trastornos neurocutáneas, puede afectar a varios tejidos del ectodermo y del mesodermo además de la piel. Las manifestaciones cutáneas predominan y son esenciales para el diagnóstico: se completan con atrofia y pigmentación lineal de la piel, herniación de la grasa a través de los defectos dérmicos y múltiples papilomas de las membranas mucosas o la piel (Lee y Goltz de 2006, Sampaio y Harper 2006). Las alteraciones dérmicas son evidentes invariablemente al nacimiento. Inicialmente se presentan lesiones rosadas o rojas, angulares y máculas atróficas; con la edad las áreas eritematosas tienden a desaparecer y las áreas de atrofia a despigmentarse. A nivel ocular (coloboma del iris y la coroides, el estrabismo y la microftalmia), (dientes hipoplásicos y papilomas de labios) por vía oral, y esquelético (sindactilia, polidactilia, deformidades campodactilia, y la ausencia de dedos) defectos, así como anormalidades neurológicas (retraso mental y diversas malformaciones cerebrales) son también características. La gravedad es variable: los resultados varían desde un mínimo de manifestaciones cutáneas potencialmente mortales a anomalías en el nacimiento. La multifacética presentaciones clínicas y la implicación de múltiples órganos vistos en este trastorno requieren el conocimiento y la experiencia de especialistas de diferentes disciplinas de la medicina.
Genética.- La HFD se trasmite de manera ligada al X dominante, siendo el 90% de los casos mujeres afectadas. El 10% de los individuos con HFD son hombres, se considera que la mayoría de los casos en hombres es letal, esto explicado por hemicigocidad del gen comprometido, se considera que el 95% de todos los casos son de novo. Muchos autores sugieren que debido a la inactivación aleatoria del cromosoma X, resulta un mosaicismo funcional explicando así la asimetría y variabilidad del cuadro clínico. Se propone como explicación a los casos reportados en hombres, la posibilidad de una mutación de media cromatide. Si la mutación ocurre tempranamente en el desarrollo embrionario, las células gonadales podrían estar comprometidas explicando de esta manera la transmisión de padres a hijos.
El gen PORNC homólogo humano de la PORCUPINA de Drosophila, (human porcupine locus MG61/PORC), localizado en el locus Xp11.23, contiene 15 exones, codifica para una O-aciltrasferasa, que cataliza la N-palmitoilación de cisteína y la O-acilación de serina en el retículo endoplasmático, necesarias para el direccionamiento de varias proteínas Wnt. Tiene 5 isoformas resultantes del splicing alternativo (exón 7 y 8) y es expresada en una amplia cantidad de tejidos. Es requerida para la secreción de algunas de las proteínas Wnt, importantes proteínas de señalización que se unen a receptores trasmembrana, estas son morfógenos indispensables en vías de señalización y estabilización intracelular, permitiendo la translocación al núcleo de β-catenina activador de genes específicos del desarrollo. La señalización Wnt es requerida para inducción, proliferación, morfogénesis de varios órganos, el PORNCse expresa en piel, ojos, oído medio, dientes, huesos largos, costillas, vértebras y lóbulo frontal del cerebro; además participa en el cambio de polaridad celular y adhesión celular.
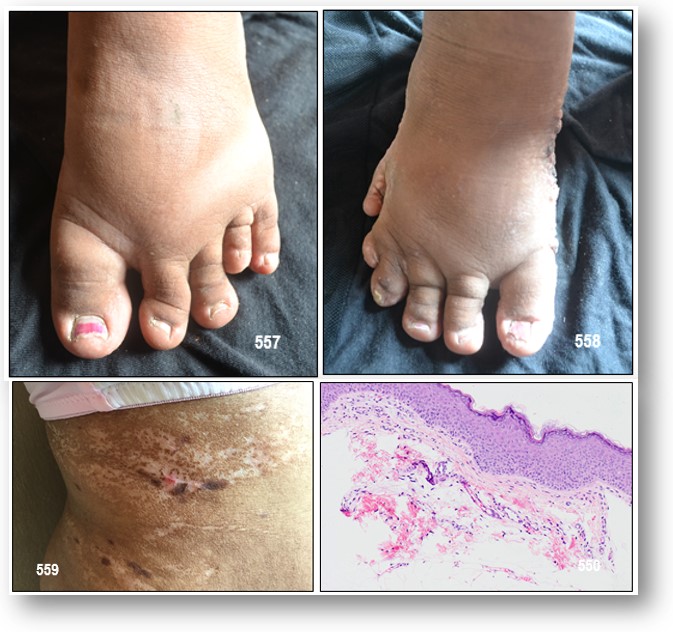
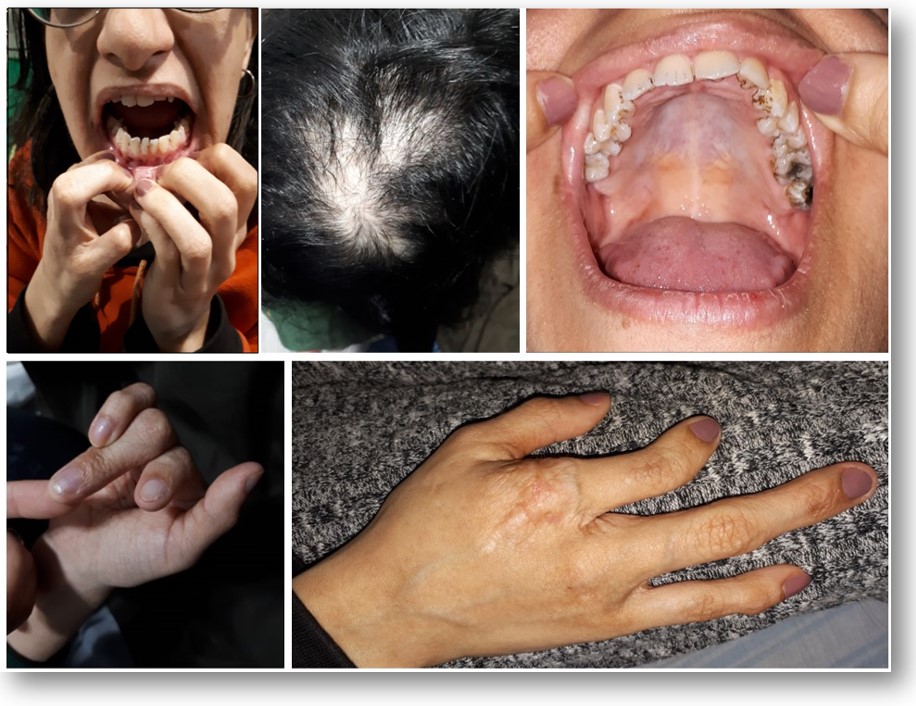
Manifestaciones Clínicas.- Las características clásicas incluyen atrofia dérmica, máculas hiperpigmentadas o hipopigmentadas, las cuales presentan un patrón asimétrico de distribución, siguiendo líneas de Blaschko. Las mismas corresponden a vías de migración neuronal durante el desarrollo embrionario y fetal de la piel. Además se encuentra herniación de tejido graso sobre defectos dérmicos, más comúnmente localizados en extremidades, particularmente en fosa poplítea y antecubital; múltiples papilomas en mucosas o piel (ojos, labios, encías, periorales y perianales), telangiectasias lineales, uñas distróficas, alopecia, en algunos casos hiperqueratosis en plantas y palmas, hipohidrosis, urticaria y fotosensibilidad. Ha sido observada en algunos casos asimetría facial, corporal y de extremidades. Otras anomalías descritas son: hipodoncia, oligodoncia, displasia del esmalte dental; microftalmia, anoftalmia, coloboma ocular, ectopia lentis, labio y paladar hendido, hemihipoplasia de lengua e hipertrofia gingival.
El compromiso óseo es variable y usualmente asimétrico, se presenta hasta en el 80% e incluye sindactilia, adactilia, ectrodactilia, polidactilia, y bradiquidactilia, ausencia o hipoplasia digital, costillas cervicales, desarrollo asimétrico de tronco y extremidades, anormalidades vertebrales, escoliosis hasta en el 20%, displasia de clavículas; seudoartrosis clavicular del lado derecho ha sido reportada en el 90% de los casos de los heredados con rasgo dominante. Raramente es bilateral. También se ha encontrado en el síndrome del puerto flotante (Floating-Harbor syndrome). La polidactilia es por lo general postaxial con clinodactilia. Hemimelia de las extremidades ha sido reportada. Otro hallazgo es la espina bífida oculta (7%), y un patrón radiológico en huesos largos muy característico, la osteopatía estriada (80%). La osteopatía estriada se considera un signo distintivo de esta enfermedad. Algunos pacientes tienen osteoporosis, displasia fibrosa y lesiones quísticas en los huesos. Han sido reportados hernia umbilical y/o onfalocele. Ha sido documentada una cola rudimentaria y un esternón divido.
A nivel del sistema genitorinario se ha reportado hidronefrosis, pelvis renal y uréteres bifidos, ectopía renal ha sido descrita hasta en el 9% de los casos reportados en la literatura, otras anomalías renales descritas fueron riñón en herradura y displasia quística. Hipoplasia de labios vaginales, y criptorquidia ha sido reportada esporádicamente. Finalmente se han descrito hipoacusia mixta, displasia coclear; infecciones respiratorias y urinarias recurrentes, sin disfunción inmunitaria asociada; hernia diafragmática, diastasis de los rectos anteriores y displacencia anal.
El desarrollo psicomotor suele ser normal, aproximadamente el 15% de los casos reportados presentan retardo mental, pero solo en aquellos casos que implican mayor severidad.
Histopatología.- Los hallazgos histológicos son característicos e incluyen dermis extremadamente delgada, por ausencia de tejido conectivo dérmico, con atenuación de las fibras de colágeno, las células grasas se extienden virtualmente a la epidermis, entremezclándose con tejido conectivo, la apariencia de la epidermis es normal, descrita como acantosis y papilomatosis. En cultivo de fibroblastos ha sido descrito alteración del crecimiento celular, con vacuolas citoplasmáticas. La microscopía electrónica muestra numerosas estructuras lineales filamentosas en la dermis, las células grasas multiloculares se observan en las lesiones grasas y son consideradas una forma de adipocitos inmaduros
La HDF puede representar un desarrollo suprimido de los fibroblastos dérmicos y/o diferenciación y proliferación preferencial de adipocitos, la señalización de las Wnt producen proliferación de fibroblastos en las heridas, sugiriendo un papel importante en el desarrollo dérmico, sin embargo, las Wnt inhiben la adipogénesis, por lo que la inhibición de la señalización podría cambiar el destino de las células mesodérmicas de fibroblastos a adipocitos. Además las proteínas Wnt inducen la diferenciación de osteoblastos a partir de las Stem Cell mesenquimales durante el proceso de osteogénesis normal; en ratones transgénicos con mutación en la vía de señalización Wnt presentan densidad ósea alterada.
La HFD es causada por la pérdida de función o deleción del gen PORNC, resultando en la falla de la señalización de las proteínas Wnt y por lo tanto en las vías de señalización en tejidos de origen ectodérmico o mesodérmico. Han sido reportadas 23 mutaciones diferentes en el gen PORNC, entre ellas missense, nonsense, deleciones, duplicaciones, microdeleciones cromosómicas y mutaciones de splicing. En algunos pacientes con síndrome de Goltz ha sido reportado deleción de la región Xp11.23, por lo cual se ha propuesto como un síndrome de genes contiguos.
La variabilidad clínica sugiere que la mutación o el nivel de inactivación del cromosoma X, se correlaciona directamente con la severidad del fenotipo; todos los casos de mujeres con deleción en PORCN muestran una extremadamente alta inactivación en X, mientras que muchas mujeres con mutación puntual tienen una inactivación aleatoria. Todos los casos reportados en hombres han sido mosaicismos para la mutación en PORCN.
Diagnóstico.- El diagnóstico se basa en los hallazgos clínicos e histológicos, además se cuenta con el estudio molecular del gen PORCN por análisis de secuenciación, detectando pequeñas deleciones o duplicaciones, inserciones, mutaciones nonsense y missense o test de grandes deleciones.
La mayor parte de los casos tiene un cariotipo normal y sucede de forma esporádica, como es el caso de nuestra paciente.
Aunque nuestra paciente no tiene afectación visceral, las manifestaciones cutáneas son lo suficientemente características como para permitir el diagnóstico de HDF.
Diagnóstico Diferencial.- El diagnóstico diferencial incluye la enfermedad de Rothmund-Thomson (lesiones eritematosas lineales induradas con fotosensibilidad, atrofia de piel y telangiectasias, la dermis es estructuralmente normal), Incontinencia pigmenti, la deleción Xp22.2, Síndrome de Adams Oliver y Aplasia cutis congénita. Respecto al nevus lipomatoso superficial de Hoffmann-Zurhelle y las poiquilodermias congénitas.
Tratamiento.- El manejo debe ser liderado por Dermatología para las lesiones de piel, tratando el dolor en los casos que este se presente, las lesiones erosivas y previniendo infecciones secundarias. Las lesiones cutáneas, especialmente las faciales tienen connotación estética, pero no representan mayor problema terapéutico; las lesiones papilomatosas generalmente son removidas quirúrgicamente, pero son recurrentes. La cirugía plástica y ortopédica temprana mejoran las deformidades en extremidades. El seguimiento por Oftalmología, Odontología y de Neurodesarrollo debe ser continuo. Por la amplia heterogeneidad fenotípica, el pronóstico y manejo dependerán de la severidad del cuadro, pacientes con leve compromiso pueden llevar una vida normal, como es el caso de nuestra segunda paciente.
En la actualidad se han reportado aproximadamente 200 casos del síndrome de Goltz en la literatura mundial indexada; en Ecuador es el primer caso reportado en literatura indexada.
Bibliografía
- Fitzpatrick, Eisen, Wolff: Dermatología en Medicina General, Editorial Médica Panamericana, Buenos Aires, Argentina, 4ª.Ed., 1997: 762-770.
- Goltz RW. Focal Dermal Hipoplasia Syndrome. Arch. Dermatol. 101:1-11,1970
- Gorlin RJ. Focal Dermal Hipoplasia Syndrome. Acta. Derma. Venereol. (Estocolmo) 43;421-440, 1963
- Jessner M. neviforme, poikilodrmie-artigehautveranderungen mit Missbildungen. Zbl. Haut Geschectisrkr. 27:46, 1928
- Zurita Salazar G, Torres M. Gaceta Dermatologica Ecuatoriana. Vol. N,1, 1992.
- Clements S, Wessagowit V, Lai-Cheong J, Arita K, Mc Grath. Focal dermal hypoplasia resulting from a new nonsense mutation, p.E300X, in the PORCN gene. J Dermatol Sci 2008; 49: 39-42.
- Wang X, Sutton VR, Peraza-Llanes J, Yu Z, Rosetta R, Kou Y-C, Eble T, Patel A, Thaller C, Fang P, Van den Veyver I. Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet 2007; 39: 836–838.
- Elliot M, Bayly R, Cole T, Temple IK, Maher ER. Clínicasl features and natural history of Beckwith Wiedemann Syndrome. Am J Med Genet 1995;56:366-73.