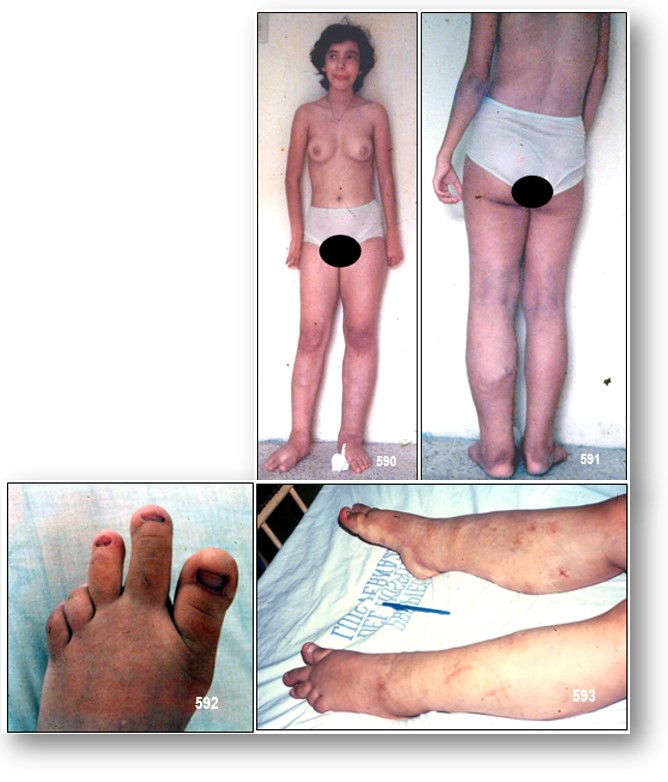

INFORMACIÓN BÁSICA: •Introducción: Síndrome de Klippel-Trénaunay (SKT).-(ORPHA:2346/CIE-10:Q87.2 /OMIM:149000-608354-608355).- El SKT es una malformación vascular combinada capilar-linfático-venosa, asociada a sobrecrecimiento óseo y/o de partes blandas, de uno o ambos miembros, o de tórax- El SKT fue descrito en el año 1900 como nevo varicoso osteohipertrófico, por la combinación de varices e hipertrofia corporal segmentaria, es considerado en la actualidad una anomalía vascular combinada de los sectores capilar, linfático y venoso. Se presenta desde el nacimiento y afecta por igual ambos sexos. Las lesiones comprometen más comúnmente uno o ambos miembros inferiores, pero también pueden presentarse en pelvis, tronco y extremidades superiores, en forma combinada con las anteriores o en forma aislada. Estas malformaciones se asocian con hipertrofia ósea o de partes blandas y drenaje venoso profundo anómalo. Las malformaciones cutáneas vasculares son múltiples y típicamente asientan en la zona lateral de alguna extremidad, nalga o tronco.
El componente es de tipo macular en el recién nacido, pero va adquiriendo un aspecto rugoso y con vesículas linfáticas, a medida que el niño crece. La hipoplasia linfática está presente en más del 50% de los pacientes y se manifiesta por linfedema o lesiones microquísticas. La vena marginal lateral, otro componente de este síndrome, se torna prominente debido a incompetencia valvular y a anomalías del sistema venoso profundo.
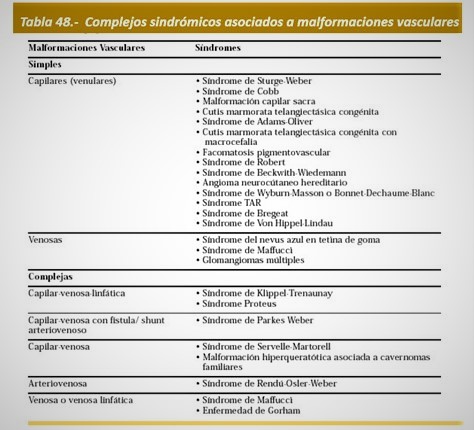
Clasificación: Según la clasificación de Mulliken y Glowacki de 1996 fue aceptada por la Sociedad Internacional para el Estudio de las Anomalías Vasculares (ISSVA), las malformaciones vasculares son un subgrupo de anomalías vasculares diferentes de los tumores y se dividen en 2 grupos: simples y combinadas. El SKT se clasifica en el grupo de las malformaciones vasculares combinadas (Tabla 44).
Etiopatogenia: Biología molecular.- La causa del SKT continúa siendo desconocida pero se postulan diversas hipótesis etiopatogénicas. Quizá la hipótesis más aceptada es la que considera que el defecto morfogenético que causa el SKT podría alterar el proceso normal de remodelación y desarrollo de estructuras vasculares (vasculogénesis y angiogénesis), por un mecanismo de interferencia de la apoptosis. Ésta se requiere en el remodelado vascular durante la embriogénesis, en la que interviene el factor antagonista angiopoyetina-II.
Los mecanismos que provocan el desarrollo de la hipertrofia están poco definidos. El sobrecrecimiento de la extremidad tiene 2 fases: aumento del volumen y el diámetro, y aumento de la longitud por crecimiento óseo. Los factores propuestos para explicar el aumento de volumen son la hipertensión venosa, producida por trombosis o atresia de las venas profundas, o que el aumento del flujo sanguíneo en los capilares y las venas anormales promueva el sobrecrecimiento de la extremidad en la vida fetal. Sin embargo, explicar el origen del crecimiento óseo es más complejo, y se ha sugerido que podría producirlo la disregulación en la formación de los canales vasculares
En cuanto al mecanismo de transmisión, el SKT se presenta esporádicamente, aunque su aparición se ha descrito en más de un miembro de una misma familia, o la presencia de malformaciones capilares o varices graves en otros miembros. En algún caso, el SKT se asocia a la presencia de hemangiomas en familiares del paciente. Estos hallazgos han llevado a algunos autores a sugerir una herencia autosómica dominante en casos concretos. Sin embargo, la distribución de los hallazgos somáticos en la mayoría de los pacientes con SKT es más compatible con una mutación somática de un gen todavía desconocido. Algunos investigadores han sugerido que el síndrome podría ser el resultado de un gen letal que sobrevive por mosaicismo. Trabajos recientes han observado que la translocación cromosómica t (5;11), que aumenta la transcripción del factor angiogénico VG5Q, y la mutación funcional E133K, que potencia el efecto de este factor, conllevan una mayor susceptibilidad a presentar el SKT. Otros autores han identificado una translocación t (8;14) (q22.3;q13) en un paciente con SKT.
Estos hallazgos son muy preliminares y requieren nuevos trabajos que permitan confirmarlos
Manifestaciones clínicas: El SKT tiene una incidencia similar en ambos sexos y está presente más o menos completamente en el momento del nacimiento. Sin embargo, la edad del diagnóstico puede retrasarse. El SKT afecta típicamente las extremidades inferiores de forma unilateral y es más frecuente en el lado derecho. También se ha descrito con menos frecuencia en ambas extremidades inferiores, en las extremidades superiores uni o bilateralmente, en las 4 extremidades simultáneamente, uni o bilateralmente, y cuadros limitados al tórax, a la pelvis o al abdomen, o sólo a la cabeza y el cuello. Entre los síntomas más relevantes están: sangrado, dolor, infección y de autoimagen negativa. El compromiso pélvico puede ser asintomático, o manifestarse por constipación, disuria, infección urinaria recurrente, hematuria micro o macroscópica y proctorragia. La tromboflebitis ocurre en 20-45% de los pacientes y también puede ocurrir tromboembolismo pulmonar.
– Malformación capilar: La malformación capilar, también conocida como mancha en vino de Oporto, nevo telangiectásico o angioma plano, es la manifestación cutánea más frecuente presente en la mayoría de los pacientes en el nacimiento. Clínicamente se presenta como una mancha rosa rojiza de bordes lineales cuya intensidad de coloración puede aumentar con la edad, y que adquiere una tonalidad más violácea (Imagen 3B). Cuando aparece en el tronco raramente cruza la línea media y presenta un borde bien definido. De aspecto macular en el recién nacido, algunos pacientes experimentan con la edad la aparición de lesiones nodulares que parecen corresponder a canales venosos o linfáticos ectásicos. En algunos casos, éstos pueden aparecer en áreas corporales no afectadas por el SKT y pueden regresar espontáneamente.
La malformación capilar se corresponde histológicamente con múltiples malformaciones vasculares contiguas formadas por capilares ectásicos o, en menor proporción, vénulas de la dermis superficial. Debido a su carácter no proliferativo, son lesiones estásicas que no regresan con el tiempo. Aunque las malformaciones capilares son la manifestación clínica más banal del síndrome, pueden presentar ocasionalmente algunas complicaciones, como infecciones, sangrado o ulceración de la piel.
Recientemente, algunos autores han clasificado las malformaciones capilares en 2 grupos: geográficas o no geográficas. Las geográficas se definen como lesiones muy bien delimitadas, de forma irregular y de color rojo oscuro o púrpura. Las no geográficas, por el contrario, presentan un límite poco preciso respecto a la piel normal en algunas áreas (no en la línea media), son de gran tamaño, con distribución segmentaria, y de color rosa suave o rojo-rosado. La presencia de manchas geográficas supone, para estos autores, un mayor riesgo de presentar malformaciones linfáticas, así como un mayor índice de complicaciones asociadas con este síndrome.
– Malformaciones venosas: Las venas varicosas atípicas, o venas laterales anómalas, y las malformaciones venosas no están siempre presentes en el nacimiento y se hacen más evidentes coincidiendo con la bipedestación del niño. En algunos pacientes con SKT, en los que la región facial está implicada, es posible que las varicosidades no sean evidentes, probablemente porque la gravedad desempeña un papel importante en su desarrollo. Las venas varicosas atípicas son venas embriológicas persistentes del sistema venoso superficial y corresponden a la vena lateral del muslo o la vena ciática. Suelen ser venas largas y tortuosas que pueden carecer de válvulas y causar síntomas de pesadez en las extremidades inferiores.
Las malformaciones venosas pueden ocurrir en los sistemas venosos superficial y profundo, e incluso en las venas perforantes de las extremidades (Imagen 2A). Entre las anormalidades en el sistema superficial, es posible hallar desde ectasia de pequeñas venas y varicosidades hasta grandes malformaciones venosas. El sistema venoso profundo suele estar afectado en más de la mitad de las malformaciones vasculares de predominio venoso de las extremidades, incluyendo a los pacientes con SKT. Su estudio minucioso, mediante ecografía-Doppler, flebografía y resonancia magnética, es fundamental cara a valorar cualquier procedimiento terapéutico.
Las alteraciones en el sistema venoso profundo incluyen dilataciones aneurismáticas, duplicaciones, hipoplasia, aplasia y compresión externa por vasos anómalos o bandas fibróticas. Las venas poplíteas y femorales superficiales son las afectadas con más frecuencia, aunque puede estar implicada cualquier vena, incluso la cava inferior. El grado de hipertensión en pacientes con obstrucción del flujo sanguíneo dependerá de la presencia de venas colaterales. Algunos pacientes también pueden presentar venas varicosas perianales y perirrectales, posiblemente debidas a un alto flujo en la vena ilíaca interna. Además, la presencia de grandes venas suprapubianas puede ser un signo de atresia de la vena ilíaca.
La afección pelviana se asocia, con frecuencia, a la presencia de una malformación capilar-linfática-venosa en la extremidad inferior y generalmente es asintomática. Ocasionalmente, las malformaciones venosas pueden extenderse al mediastino posterior y al espacio retropleural, aunque raramente producen síntomas.
En las malformaciones vasculares venosas o mixtas extensas se ha descrito una coagulación intravascular localizada hasta en el 88% de los pacientes.
Este trastorno, en función de la gravedad, puede producir hemorragias y trombosis en situación basal y empeorar claramente con algunos tratamientos (inmovilización posquirúrgica, escleroterapia). Para su estudio se recomienda realizar un hemograma, donde el número de plaquetas suele estar en el límite bajo de la normalidad (distinto de la coagulación intravascular diseminada [CID] que se produce por atrapamiento local de plaquetas en el fenómeno de Kasabach-Merritt, medir el tiempo de protrombina, y valorar el fibrinógeno (que suele estar disminuido), el dímero-D (habitualmente elevado) y la presencia de complejos solubles de fibrina (que suelen estar presentes).
Las complicaciones que se pueden asociar a la presencia de varices o malformaciones venosas son la hemorragia, la tromboflebitis en un 20-45% de los pacientes, la presencia de comunicaciones arteriovenosas, que pueden acompañarse de fallo cardíaco congestivo por alto flujo, y la tromboembolia pulmonar en un 4-25% de los pacientes. Las complicaciones pelvianas van desde la presencia de hematuria, hemorragia gastrointestinal y estreñimiento, hasta la obstrucción del meato vesical y las infecciones recurrentes por flora intestinal.
–Malformaciones linfáticas: Pueden deberse a una hipoplasia linfática, presente en más del 50% de los pacientes, y asociarse a linfedema y/o macroquistes linfáticos aislados en la pelvis y las extremidades superiores, o microquistes en la pared abdominal, la región glútea y la zona distal de las extremidades. Las malformaciones linfáticas suelen ser predominantemente subcutáneas o pueden infiltrar difusamente el músculo, y su presencia se asocia a un mayor número de complicaciones (véase la Tabla 49).

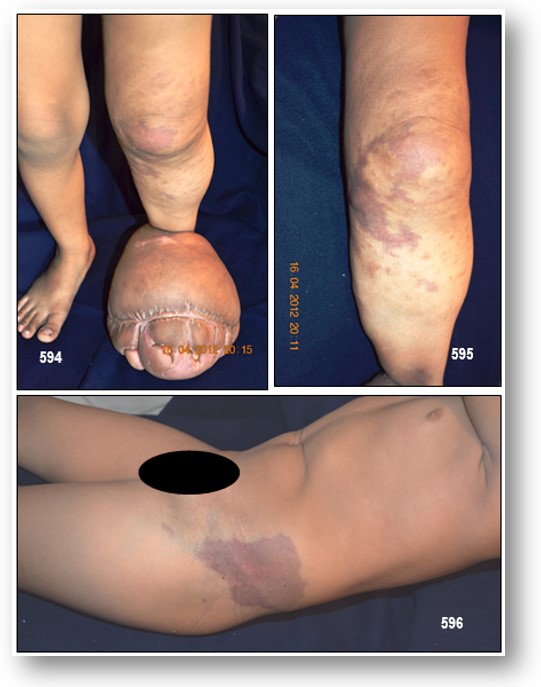
-Alteración osteoarticular: La hipertrofia es el rasgo más variable del SKT, generalmente presente en el momento del nacimiento. El aumento de tamaño de la extremidad puede ser únicamente circunferencial o presentar, además, un incremento en su longitud. No es posible predecir la progresión que presentará una extremidad hipertrófica, pero generalmente no se producen grandes cambios tras el nacimiento, sino más bien un crecimiento axial lentamente progresivo. En algunos pacientes con SKT, la extremidad afectada es corta o hipotrófica. Entre las complicaciones de la hipertrofia se encuentra la presencia de celulitis de repetición, generalmente asociada a linfedema, y la existencia de discrepancia en la longitud de ambas extremidades.
Otras complicaciones que se pueden asociar al SKT, entre las que destacan las úlceras y los cambios tróficos cutáneos.
Los pacientes con SKT pueden presentar, además, otros defectos congénitos asociados que se enumeran en la tabla 2.
Diagnóstico: El SKT, como ya se ha comentado al inicio de este artículo, se define como la asociación de una malformación capilar, hipertrofia de tejidos blandos y/o óseos, y la presencia de venas varicosas o malformaciones venosas, a menudo con venas laterales embrionarias persistentes. El diagnóstico del SKT es posible con la presencia únicamente de 2 de los 3 rasgos clínicos anteriores.

Para realizar una correcta evaluación del paciente con SKT, tras un exhaustivo examen clínico, muchas veces es necesario recurrir a una serie de procedimientos de imagen, tratando de utilizar, siempre que sea posible, los menos invasivos.
– Radiografía simple: Permite medir la longitud de las extremidades y detectar si existe una discrepancia entre ambas. Clínicamente sólo es posible detectar diferencias de longitud entre 0,5 y 1 cm, por lo que para valorar discrepancias menores se requiere el empleo de estos métodos de imagen. Por otro lado, se puede determinar la tasa de crecimiento de cada extremidad, conocer si las discrepancias son progresivas o estables y determinar el momento más adecuado para realizar los procedimientos terapéuticos necesarios a fin de igualar la longitud de las extremidades, datos que no es posible valorar sólo clínicamente.
La realización de radiografías no suele ser necesaria antes de los 2-3 años de edad ni más allá de la madurez esquelética, ya que las diferencias en longitud.de las extremidades no progresan tras el cierre de los cartílagos fisarios. Por ello, se recomienda realizar una medición clínica y radiológica inicial a los 2 años de edad y repetir anualmente estas determinaciones.
– Ecografía-Doppler: Antes de realizar cualquier procedimiento destinado a la eliminación de las venas varicosas superficiales, es imprescindible asegurar la presencia de un sistema venoso profundo permeable. La ecografía-Doppler suele ser el procedimiento utilizado de primera elección y, en ocasiones, el único necesario, ya que es un método de realización sencilla, sin riesgos para el paciente. Sin embargo, no permite evaluar tan nítidamente la mitad inferior de las piernas, por lo que en algunos casos se requerirá la realización posterior de una flebografía.
– Flebografía: La flebografía ascendente consiste en la a inyección de contraste yodado a través de una vena superficial del dorso del pie, para realizar posteriormente radiografías de dicha extremidad. Mediante diferentes maniobras de compresión permite estudiar el sistema venoso profundo. También resulta de utilidad para conocer la anatomía de las venas superficiales, el estado de las válvulas, la presencia de venas embrionarias, las conexiones entre los sistemas superficial y profundo, y el grado de incompetencia venosa. La flebografía tiene como desventajas la posibilidad de que se produzcan reacciones alérgicas y trombosis venosas debidas al uso de contraste yodado. Además, puede ser un procedimiento difícil que, en ocasiones, requiere múltiples inyecciones y maniobras compresivas.
En los casos en que la flebografía esté contraindicada o no permita valorar las estructuras vasculares con claridad, es posible realizar una angioresonancia con gadolinio.
– Angioresonancia: Este procedimiento se realiza mediante la inyección de gadolinio y la realización posterior de una resonancia magnética en la zona que se pretende estudiar.
Permite documentar el tipo y la distribución de las anomalías vasculares y la anatomía del drenaje venoso. Es una técnica de gran utilidad para la identificación de la patognomónica vena marginal de Servelle, presente en el tejido subcutáneo de la pantorrilla y el muslo. Además, la angioresonancia aporta información sobre la presencia o ausencia de afección vascular intraabdominal y pelviana, así como sobre su extensión. Tiene la desventaja de ser un procedimiento más costoso y de que para su realización requiere una resonancia.
Por otra parte, la resonancia magnética clásica es la técnica de elección para la valoración de las partes blandas. En los pacientes con SKT es útil para conocer la naturaleza de la hipertrofia (tejido subcutáneo, músculo y/o hueso), medirla y compararla con la extremidad contralateral.
Además, permite detectar y conocer la extensión y la infiltración de las malformaciones venosas y linfáticas macro y microquísticas, y evaluar la pelvis o las estructuras craneales que no se pueden estudiar fácilmente con ecografía.
– Arteriografía: Puede ser una prueba necesaria en casos aislados en los que se sospeche la presencia de fístulas arteriovenosas. La existencia de grandes fístulas arteriovenosas no es típica del SKT y sugiriere el diagnóstico de síndrome de Parkes-Weber. Sin embargo, la presencia de fístulas microarteriovenosas puede manifestarse en el SKT.
– Linfoescintigrafía: Aporta información sobre el drenaje linfático y puede estar indicada en algunos pacientes para la valoración de los vasos linfáticos, en caso de linfedema persistente o cuando las diferencias en el diámetro de ambas extremidades superen los 4 cm. La ecografía es el método de elección para establecer un diagnóstico prenatal del SKT, por la observación de una hipertrofia o una asimetría en las extremidades, o bien de una masa vascular.
Diagnóstico diferencial: El diagnóstico diferencial del SKT se establece con otra serie de síndromes que comparten alguno o algunos de los rasgos clínicos del SKT, y estos son:
- – Síndrome de Proteus. Síndrome de Maffucci.
- – Síndrome de Bannayan-Riley-Ruvalcaba.
- – Síndrome de Parkes-Weber. Síndrome de
- – Sturge-Weber y la Macrodactilia. El SKT debe diferenciarse de otras causas de macrodactilia, como el síndrome de Proteus, la neurofibromatosis tipio 1 y la enfermedad de Milroy.
Tratamiento: El manejo de estos pacientes debe ser multidisciplinario, y su tratamiento es fundamentalmente clínico y conservador. Idealmente deberían ser controlados anualmente. Si surge una discrepancia de miembros superior a 1.5 cm. (tal como presenta mi paciente pediátrica), se deberá indicar un resalto en el miembro contralateral más corto, para prevenir claudicación y escoliosis secundaria. Son recomendables las medias de compresión elástica a medida. El agrandamiento grosero del pie requiere procedimientos ablativos selectivos, que le permitan al niño usar zapatos adecuados. En pacientes seleccionados, la escleroterapia puede ser usada para obliterar venas superficiales incompetentes o para achicar malformaciones vasculares.
Bibliografía
- Ricardo García Mónaco, Tamara Kreindel, Ana Giachetti. Malformaciones vasculares: claves diagnósticas para el radiólogo. RAR – Volumen 76 – Número 4 – 2012
- Fitzpatrick´s Dermatology in General Medicine. Seventh edition. Edited by Wolf; Goldsmith, Katz, Gilchrest, Paller and Leffell. Mc Graw Hill medical publishing división. 2007, chapter 136, 1281-88.
- Belov, S.: Classification of congenital vascular defects. Int Angiol 1990; 9: 141-146.
- Finn, M.C.; Glowacki, J.; Mulliken, J.B.: Congenital vascular lesions: clínicasl application of a new classification. J Pediatr Surg 1983; 18: 894-900.
- Kelly M. Kassabach-Merritt Phenomenon. Pediatr Clin N Am 2010; 57: 1085-1089.
- Dompmartin A, Acher A, Thibon P, Tourbach S, Hermans C, et. Al. Association of Localized Intravascular Coagulophaty with Venous Malformations. Arch Dermatol 2008; 144(7): 873-877. 2. Aguado Gil L, Redondo Bellón P. Utilidad de los marcadores angiogénicos y protrombóticos en la toma de decisiones terapéuticas en pacientes con malformaciones vasculares. Piel. 2011. doi: 10.1016/j.piel. 2010.12.016.
- Spring MA, Bentz ML: Vascular anomalies. En: Bentz ML, Bauer BS, Zuker RM. Principles & practice of pediatric plastic surgery. Quality Medical Publishing Inc.St. Louis, Missouri, 2ª ed. 2008. Pp:127
- R.V. Moreno Martín, P. Martínez Brándulas, R. Palazón García, S. Gil Hernández. Síndrome de Klippel-Trénaunay: a propósito de un caso. Rehabilitacion 2004;38:188-91 – DOI: 10.1016/S0048-7120(04)73455-6.
- Gimeno Pita, P. Pérez Martín, J. López-Pisón, M. Romeo Ulecia,N. Galeano Riaño,M. Marco Tello. Síndrome de Klippel-Trénaunay: a propósito de tres nuevas observaciones. An Esp Pediatr, 53 (2000), pp. 350-354.
- Aelvoet, P. Jorens, L. Roelen. Genetics aspects of the Klippel-Trénaunay syndrome. Br J Dermatol, 126 (1992), pp. 603-607.