INFORMACIÓN BÁSICA: Esclerosis Tuberosa (ET) o Complejo Esclerosis Tuberosa (ET) (OMIM:191-100/ 613254/CIE-10: Q85.1/ORPHANET:805).-La ET es una facomatosis. Estas constituyen un grupo heterogéneo de enfermedades genéticas, también conocidas como síndromes neurocutáneos, que se caracterizan por anomalías en tejidos neuroectodérmicos y por padecer una predisposición incrementada para neoplasias neurológicas y cutáneas. Describiré tres de las cuatro facomatosis conocidas.
La ET un trastorno multisistémico que afecta a la piel, sistema nervioso central, riñones, corazón, huesos, ojos y otros órganos (Pascual-Castroviejo et al; 2006).
Los angiomiolipomas, astrocitomas de células gigantes, las complicaciones cardiacas y pulmonares son las principales causas de morbimortalidad (Curatolo et al; 2008). Algunas presentaciones de la ET como espasmos infantiles asociados a manchas hipocrómicas son fácilmente reconocibles y conducen al diagnóstico. Otras presentaciones son totalmente asintomáticas como ocurre con las lesiones óseas. Sin embargo también hay manifestaciones clínicas que aparecen, crecen y desaparecen con el tiempo como los rabdomiomas cardíacos y los quistes renales (Staley et al; 2011).
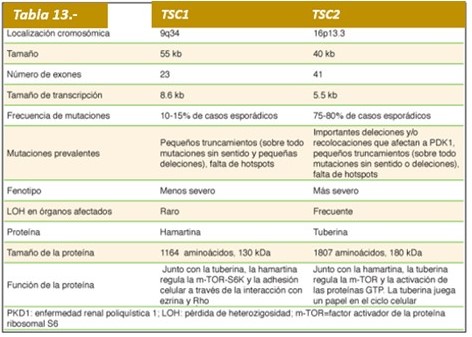
Esta entidad fue descrita por Bourneville en 1880, es una enfermedad heredada en forma autosómica dominante, con alta penetrancia y expresividad fenotípica muy variable. La ET es causada por mutaciones en 2 genes (TSC1 y TSC2). Algunos estudios reportan que alrededor de la mitad de los casos de familias con ET, tienen afectación en el locus génico 9q 34 y otros en el 11.6P 13; se implican también los locus 11q 21. Si se afecta uno de los genes puede ocurrir la enfermedad. El gen TSC1, mencionado anteriormente, se encuentra en el cromosoma 9 y da lugar a una proteína llamada hamartina. A diferencia del gen TSC2, que se encuentra situado en el cromosoma 16 y causa la proteína llamada tuberina. El gen TSC1 fue hallado en el 1997 y el gen TSC2 se descubrió en el año 1993. Los científicos opinan que estas proteínas (hamartina y tuberina) intervienen como supresores del crecimiento del tumor, regular la diferenciación celular y los procesos de proliferación. En estos procesos las células nerviosas se parten para dar lugar a las nuevas generaciones de células y se obtienen características individuales.
Datos importantes: Los síntomas de la ET pueden presentarse en el momento de nacer. Aunque en algunos pacientes el avance de los síntomas pueden presentarse más tardes. Existe variabilidad en el grado de la enfermedad, es decir, algunos pacientes presentan una forma leve de la enfermedad, otros pueden presentar discapacidades severas.
En casos excepcionales, las masas anormales pueden poner en peligro la vida. No se necesita a los dos padres para transmitirse la mutación, con un solo miembro es suficiente para que el niño consiga la enfermedad.
Aun así, en la mayoría de los pacientes con ET, se produce por nuevas mutaciones, por lo que generalmente no existe un antecedente familiar de la enfermedad, por lo que la enfermedad se obtiene a través de un proceso llamado mosaicismo gonadal (la mutación afecta a una parte de los gametos: óvulos o espermatozoides). En los últimos años, se ha detectado que el gen de la ET está enlazado al lugar de grupo sanguíneo ABO y al encogen c-abl (situados, los dos, en el brazo largo del cromosoma 9 (9q 34)). Se necesitan más estudios para confirmar más locus genéticos, algunos ya evidenciados: 11q 14-23, 16q13.3.
Epidemiología: La ET se da en personas de diferentes grupos étnicos y de ambos sexos. A nivel mundial, se afirma que hay cerca de 1 a 2 millones de individuos, y se cree que incide en 1 de cada 6000 recién nacidos. En EEUU, existirían entre 25 000 y 40 000 casos. La incidencia se ha calculado en menos de 1 caso por 100.000 persona/año.
Sintomatología:
-Manifestaciones cutáneas y mucosas:
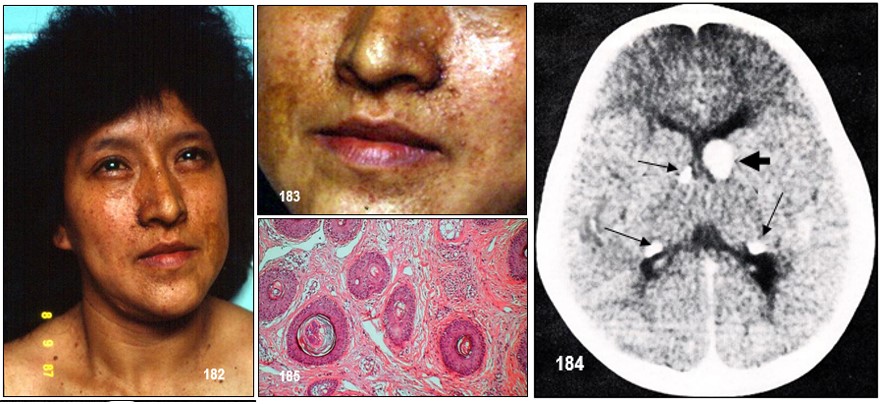
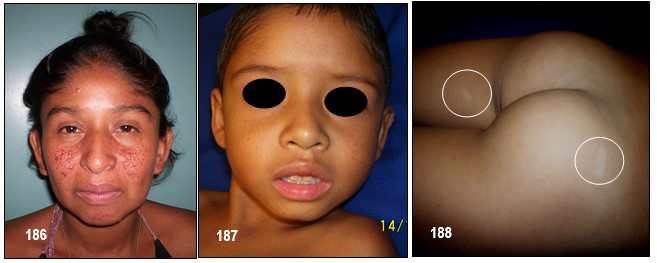
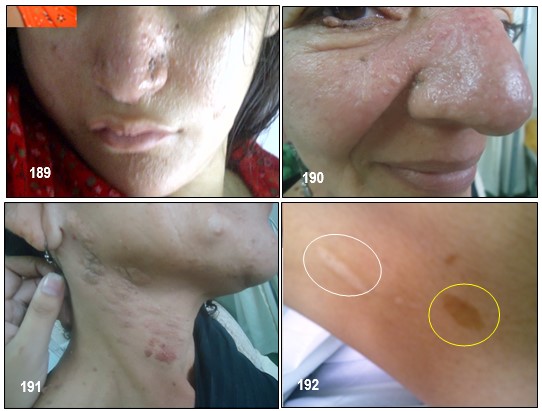
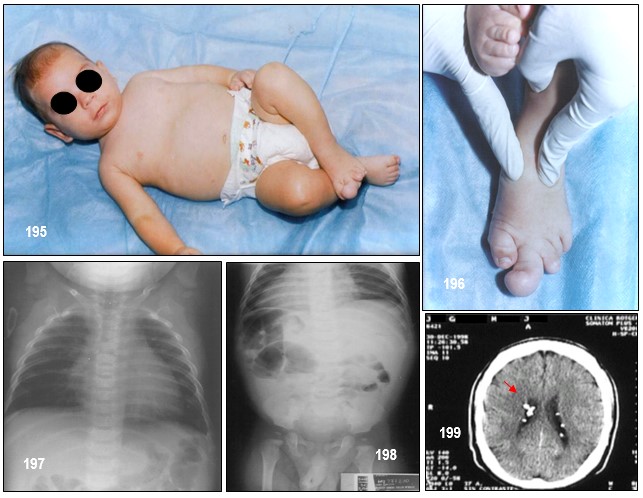
– Manchas hipocrómicas o acrómicas: Se trata de unas manchas que aparecen en la piel, en forma de hoja lanceolada o en hoja de fresno (véase la imagen 192). Las lesiones cutáneas presentes en el CET incluyen manchas hipocrómicas, placa de chagrín, fibromas ungueales ( tumores de Köenen; véase imagen 19 con fines didácticos) y angiofibromas faciales (imagen 182,186). El 90% de los pacientes presentan una o más de estas lesiones, pero ninguna es patognomónica (Roach, Sparagana; 2004). Las lesiones cutáneas son edad-dependientes (Staley et al; 2011).
Las manchas hipocrómicas son la manifestación cutánea más temprana del CET, apareciendo en un 90% de los lactantes menores de dos años (Díaz el al; 2010). Normalmente están presentes al nacimiento, pero a menudo son difíciles de visualizar en el recién nacido sin luz ultravioleta (Roach, Sparagana; 2004). Generalmente son múltiples, de bordes irregulares y forma lanceolada, de 1 a 3 cm, distribuyéndose en tronco y extremidades (Díaz el al; 2010). La mejor manera de verlas es bajo la luz ultravioleta (lámpara de Wood) (Curatolo et al; 2008). También pueden aparecer lesiones en “confeti”, que son otro tipo de máculas hipocrómicas que se asocian al CET, tienen forma poligonal y se distribuyen simétricamente sobre las extremidades, suelen aparecer en la segunda década de la vida. Puede haber afectación de áreas de pelo terminal despigmentadas, que a lo que se denomina poliosis (en el cabello, cejas y pestañas) (Díaz el al, 2010; Roach, Sparagana 2004).
Las manchas hipocrómicas no son especificas de CET porque individuos normales pueden presentar 1 ó 2 manchas hipocrómicas (Roach, Sparagana; 2004).
– Placa de chagrin (placa en piel de zapa): es un hamartoma cutáneo. Según las series varía entre el 21-80%. Aparecen alrededor de la pubertad, pero pueden pasar desapercibidas dado que suelen tener un diámetro menor de 3 cm. Son áreas de piel ligeramente elevada, amarillenta, situadas normalmente en la región lumbosacra, rodeadas de lesiones más pequeñas.
Histopatológicamente la dermis está ocupada por un colágeno denso y esclerótico con disminución e incluso desaparición de las fibras elásticas (Monteagudo et al; 2007). La frecuencia de esta lesión aumenta con la edad (Curatolo et al; 2008).
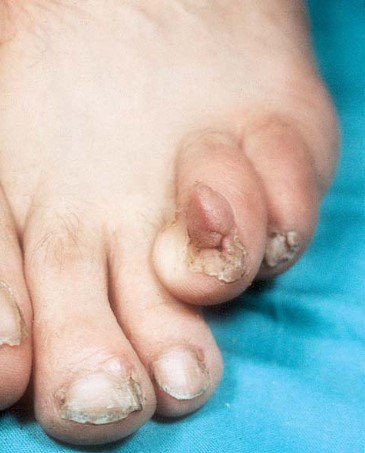
Los fibromas ungueales (tumores de Köenen) (Curatolo et al; 2008) se presentan en la adolescencia, son excrecencias firmas, lisas de color de la piel normal, que emergen de los pliegues ungueales. El tamaño es variable, miden entre 5-10mm. Aparecen en el 20% de los pacientes con CET (Díaz el al, 2010; Roach, Sparagana 2004). Pueden ser inducidos también por un traumatismo, en cuyo caso no se considera criterio diagnóstico de CET (Curatolo et al; 2008).
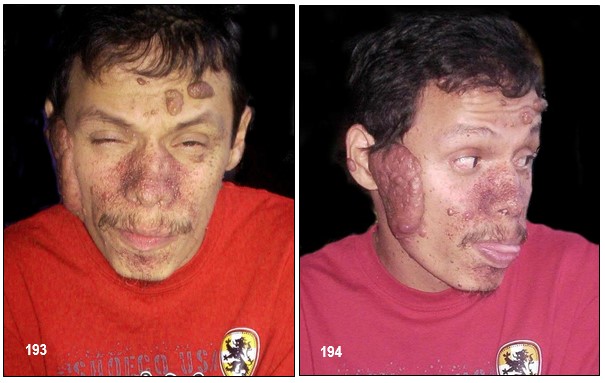
– Angiofibromas o adenomas faciales: suelen aparecen en la edad preescolar (2-5 años) (Curatolo et al, 2008; Díaz el al, 2010). Se encuentran hasta en el 80% de los pacientes mayores de 5 años (Curatolo et al; 2008), y suelen crecer rápidamente en la pubertad (Staley et al; 2011). Son pápulas firmes, rojas o marrones. Se extienden desde los pliegues nasolabiales a las mejillas y barbilla, y ocasionalmente en las orejas. En su interior poseen tejido conectivo y vascular (Curatolo et al, 2008; Roach, Sparagana 2004). Normalmente son bilaterales (Grajkowska et al; 2010). Estas lesiones también aparecen en individuos con neoplasias endocrinas múltiples tipo I (Roach, Sparagana; 2004).
– Placas fibrosas frontales: suelen aparecer en la infancia temprana, aunque pueden verse a cualquier edad (Díaz el al; 2010), se han descrito casos de desarrollo en el periodo neonatal, y a veces, puede ser la primera manifestación cutánea del CET (Curatolo et al; 2008). Es una placa de color amarillo-marrón o rojo, sobre elevada, que es variable en forma y tamaño (desde milímetros a varios centímetros) (Curatolo et al; 2008).
– Piel de naranja: Con una textura irregular como la naranja, generalmente en el dorsal o lumbar.
Manifestaciones neurológicas:
- – Crisis epilépticas: Alrededor del segundo año de vida en un 80-90% de los casos. Si se aparece tempranamente, suele cursarse en forma de síndrome de West o de Lennox-Gastaut, en general asociado a mayor compromiso cognitivo y conductual.
- -Trastornos de la conducta: Trastornos mentales, trastornos del comportamiento o alteraciones psicóticas, pueden asociarse a la esclerosis tuberosa. Las manifestaciones neurológicas de la ET se manifiestan por crisis epilépticas, retraso mental, trastornos de conducta y astrocitomas cerebrales. Diversos autores han reportado que entre el 17% y el 61% de los pacientes padecen autismo, y que entre el 0,04 y 9% de los autistas tienen esclerosis tuberosa.
Otras manifestaciones: Astrocitomas de células gigantes intraventriculares adyacentes al forámen de Monro (los tumores ubicados en Monro tienden a crecer y se necesitan controles anuales para evaluar cirugía, el ritmo de crecimiento varía aún en un mismo paciente) las posibilidades de recidiva están presentes, pero va disminuyendo hacia la adolescencia hasta calcificarse).
Los síntomas pueden ser tempranos o tardíos, dependiendo de la herencia del paciente, entre éstos puede presentarse con convulsiones, mareos, trastornos del sueño, presión intracraneal por obstrucción, presión ocular.
Calcificaciones que no crecen, pero juegan un papel fundamental en las convulsiones al funcionar como pilas, (que al descargarse causan crisis o ser un factor que empeore una epilepsia preexistente) y en el comportamiento del paciente dependiendo del área del cerebro.
Manifestaciones oftalmológicas: Las lesiones en la retina son las mas frecuentes y están presentes hasta en el 87% de los pacientes con ET (Roach, Sparagana; 2004), manifestándose con áreas acrómicas, astrocitomas y hamartomas retinianos.
Los hamartomas de la retina se encuentran entre 40-50%. Se pueden ver a cualquier edad, se han descrito casos en niños pequeños incluso en recién nacidos (Curatolo et al; 2008).
Existen distintos tipos morfológicos de hamartomas. El más común es una lesión que es relativamente plana, de superficie lisa, de color salmón a salmón-grisácea, semitransparente y de forma circular y oval, normalmente localizada en la parte superficial de la retina, cerca del polo posterior (Curatolo et al; 2008).
El segundo tipo de hamartoma es la clásica lesión multinodular elevada, que se reconoce con facilidad porque se asemeja a moras. Estos tumores se caracterizan por racimos de pequeños gránulos, quistes o nódulos brillantes. Se localizan cerca o en el margen del disco de la retina, aunque también pueden observarse en el centro de la retina periférica. Son frecuentemente bilaterales. Suelen ser asintomáticos, excepto si se localizan en la mácula o el nervio óptico. Aparecen el 50% de los pacientes con ET (Curatolo et al; 2008).
El tercer tipo de hamartoma se denomina tumor transicional o mixto, porque presenta características de los dos anteriores.
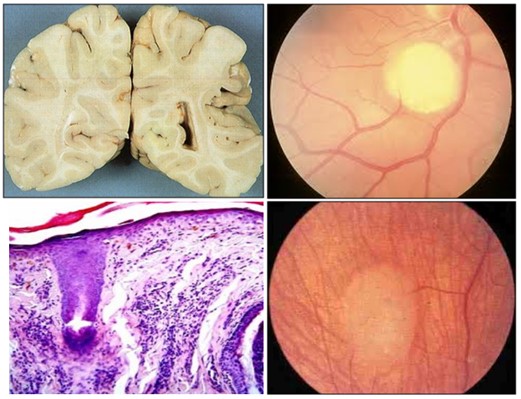
Otras lesiones que podemos encontrar a nivel de la retina son lesiones despigmentadas de la retina que parecen una sección en “sacabocados” (Roach, Sparagana; 2004).
La pérdida visual puede asociarse con hamartomas de la retina, puede desarrollarse a partir de la implicación del nervio óptico o retina. También podemos encontrar hemorragia vítrea o defectos pigmentarios del iris (Roach, Sparagana; 2004). En pacientes con TSC1 no hay lesiones en retina.
Lesiones viscerales
Renales
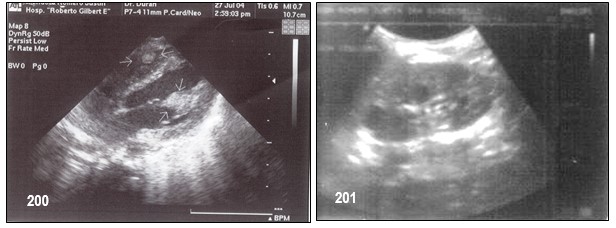
- – Angiomiolipomas: son tumores benignos compuestos por tejido vascular, muscular y graso. En individuos con CET suelen aparecer en ambos riñones de forma múltiple. Su crecimiento y la alteración de la función renal son asintomáticos si se desconoce el cuadro. Pueden presentarse cólicos, hematuria, repentina baja de presión arterial y fatiga cuando ya representa un problema quirúrgico, que requieran nefrectomía total o parcial. Si mantiene buena funcionalidad renal, cuando los angiomiolipomas son mayores a 6 cm se puede optar por una embolización selectiva.
La ruptura súbita de un angiomiolipoma puede suceder en cualquier momento (llamado Síndrome de Wunderlich) causando una descompensación renal, ya que la sangre dentro del angiomiolipoma inunda el resto del riñón, requiriendo asistencia médica de urgencia. Quistes únicos o múltiples; pueden aparecer junto a los angiomiolipomas y tienen probabilidades de malignizarse.
Cardíacas
- – Rabdomioma o hamartoma cardíaco único o múltiples se encuentran al nacer, pueden causar obstrucción ventricular, de origen mayoritariamente benignos, van reabsorbiéndose con el paso del tiempo sin dejar secuelas generalmente, el tamaño de éstos pueden variar.
Pulmonares
- – Poliquistosis: suele ser la más común.
- – Linfangioleiomatosis pulmonar: se presenta en ET con una incidencia mayor en mujeres que en hombres después de los 30 años. Se estudia una implicancia crónica de origen hormonal (hiperparatiroidismo).
Esta enfermedad se genera tejido linfático en los alvéolos pulmonares, obstruyéndolos, causando una disminución de la capacidad respiratoria progresiva, con cirugía por trasplante o desenlace fatal en algunos casos. Dentro de los síntomas puede haber: cansancio, disminución del oxígeno en sangre, entre otras.
Vasculares
- – Displasias. Algunas de éstas pueden ser aneurismas cerebrales.
- – Lesiones esqueléticas
- – Las lesiones óseas
Suelen ser geodas de 1 a 3 mm, seudoquísticas, metacarpianas y metartasianas o falángicas, o bien zonas de hiperostosis.
Manifestaciones hepáticas.- Se han encontrado AMLs hepáticos en el 16-24% de pacientes con ET. Son múltiples y normalmente asintomáticos. Más frecuentes en mujeres adultas (Curatolo et al; 2008).
Manifestaciones orales
- – Placas fibrosas ubicadas en la zona de las encías, labios y lengua.
- – Lesiones esqueléticas
- – Paladar hundido, labio fisurado y la hiperostosis.
Otras
- – Tumor odontogénico calcificado, fibroma desmoplástico, hemangiomas mucosos y/o intraóseos, mixoma odontogénico, asimetría facial, úvula bífida, retraso de la erupción y diastemas, entre otras.
Diagnóstico: En el 2012, el Consenso Internacional de CET publicó los criterios de diagnóstico de la ET (tabla 14).
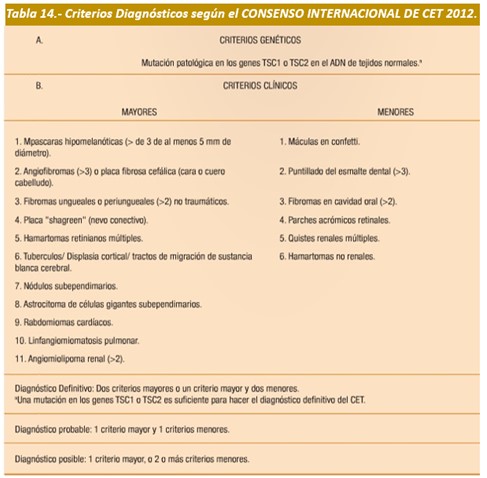
Para la aparición de CET se requiere que uno de los 2 alelos de TSC1 o TSC2 esté inactivo, sin embargo en algunos tejidos además se produce inactivación del segundo alelo (pérdida de heterocigocidad). Existe mayor evidencia de pérdida de heterocigocidad en AML renales y menor en astrocitomas subependimarios de células gigantes (SEGA por su sigla en inglés). Hay tumores en los que no se ha observado pérdida de heterocigocidad (túberes corticales y fibromas ungueales), lo que sugiere la existencia de otros mecanismos involucrados como por ejemplo haploinsuficiencia. Las proteínas hamartina y tuberina forman un dímero citoplasmático que actúa a través de la proteína Ras homolog enhanced in brain (Rheb), que regula positivamente su actividad GTPasa y disminuye la estimulación de mTOR. mTOR es una serina/treonina quinasa que cumple funciones reguladoras centrales de muchas vías de señalización, incluyendo regulación de proliferación, tamaño/crecimiento celular, traducción, metabolismo, autofagia, angiogénesis y supervivencia en respuesta a la disponibilidad de nutrientes (glucosa y aminoácidos). Varios estudios que han analizado la relación entre genotipo y fenotipo han descrito que los pacientes con mutaciones del gen TSC2 presentan una evolución con síntomas más severos.
Tratamientos: Debido a la gran variedad de síntomas, y del amplio espectro que éstos pueden presentar, no existe un tratamiento específico para esta enfermedad. Así que el tratamiento se basa en tratar cada síntoma que presente la persona afectada.
– Síntomas cutáneos: Angiofibromas: Son adenomas sebáceos que se pueden eliminar con láser. También puede reiterarse su uso con las sucesivas recurrencias. Los angiofibromas faciales formados de tejido vascular y conectivo se presentan generalmente antes de los 10 años. Los fibromas ungueales son lesiones carnosas originadas debajo de las uñas.
Los pacientes con ET pueden presentar otras manifestaciones como pólipos hamartomatosos rectales, lesiones en ovarios, hamartomas tiroideos, etc.
- – Síntomas neurológicos: Crisis epilépticas: Se utilizarán medicamentos antiepilépticos según prescriba el médico a fin de intentar controlar las crisis (ácido valproico, carbamazepina, etc.) aunque ello no garantiza el control de éstas.
- – Retraso mental: Se puede necesitar la ayuda de educación especial dependiendo del grado de retraso.
- – Síntomas viscerales y esqueléticos: La actuación dependerá de la gravedad, localización y sintomatología de la lesión. Así, por ejemplo, el rabdomiomas no suelen dar síntomas y suelen desparecer con la pubertad, así que no es necesaria su extirpación. Otras afectaciones viscerales, como los quistes renal o pulmonar, aneurismas, tumores u otros requerirán tratamiento específico definido.
Desde la aparición de la enfermedad, se tienen que tomar una serie de tratamientos de estimulación precoz (psicomotricidad, fisioterapia, logopedia…) resaltar que a lo largo de su vida necesitará apoyo en lecto-escritura y practicar natación para fortalecer los músculos por la hipotonía que producen las crisis epilépticas.
En las diversas patologías, a parte de las crisis epilépticas, la más grave es el problema de conducta, por ello también es necesario apoyo psicológico para la orientación de sus familiares.
A lo largo de la evolución se realizarán una serie de intervenciones, electroencefalograma si hay crisis epilépticas, su periodicidad dependerá del grado de las crisis. TAC craneal cada cinco años, para un correcto control de los nódulos subependimarios y de su localización en relación al agujero de Monro. RM cerebral, en caso de que se plantee la exéresis quirúrgica de algún túber cerebral cortical, pues esta exploración define mejor las estructuras cerebrales que el TAC. Y por último, se hará psicometría y cuantificación del coeficiente intelectual, especialmente en niños con problemas escolares o en el momento de comenzar la escuela, para situarles en el nivel educativo adecuado. Es preciso efectuar una vigilancia con práctica de controles periódicos, para detectar precozmente la aparición de complicaciones tumorales. Se ha señalado que la edad media del fallecimiento de estos pacientes se situaba en torno a los 24 años (Webb y Cols, 1996) pero otros estudios (Jancar, 1996) indican que su longevidad se ha incrementado en los últimos tiempos.
Evolución: La evolución de la ET tiene una tendencia de progreso conforme el afectado va avanzando en edad, al igual que van aumentando las alteraciones existentes. Según el órgano afectado del individuo, tendrá una evolución u otra y la edad de fallecimiento va en relación con el tamaño de los tumores. El pulmón, el riñón y el sistema nervioso central son los que determinan el pronóstico.
Prevención: El tratamiento más efectivo de esta entidad es su prevención. En caso de antecedentes familiares se recomienda el consejo genético. Si uno de los padres se encuentra afectado, la posibilidad de transmitir la enfermedad se estima en el 50% se debería advertir a la pareja del riesgo que conlleva la procreación mas no desaconsejarla, debido a que esto solo es decisión de la pareja.
Existe también la disponibilidad de diagnóstico prenatal de mutaciones conocidas en el ADN, pero ya que la enfermedad aparece frecuentemente como mutaciones nuevas, raramente se puede prevenir.
Comentarios de los Editores: El complejo esclerosis tuberosa (CET) es una enfermedad autosómica dominante, de expresividad variable y afectación multisistémica, caracterizada por el crecimiento de tumores benignos denominados hamartomas. Los órganos principalmente afectados son el cerebro, la piel, el riñón, el ojo, el corazón y el pulmón. Un reciente artículo aborda la demostración de que, en el CET, mutaciones en los genes TSC1 yTSC2 alteran la cascada enzimática mTOR (mammalian target of rapamycin), que regula múltiples funciones, como la síntesis proteica y la plasticidad sináptica. En la actualidad, se valora la posibilidad de recurrir a tratamiento con inhibidores de la vía mTOR (sirolimus, everolimus, tensirolimus). El 85% de los niños con CET presenta manifestaciones neurológicas que, por su gravedad, constituyen la mayor causa de morbimortalidad. Las principales manifestaciones neurológicas son la epilepsia, los trastornos del espectro autista y el retraso mental.
Bibliografía
- Northrup H, Koenig MK, Au KS. Tuberous sclerosis complex. In: Pagon RA, Bird TD, Dolan CR, et al., eds.Gene Review
- Enfermedades neurocutáneas, En: Fitzpatrick TB, Eisen A, Wolgg K, Freednerg IM, Austen KF, editores. Dermatología en Medicina General. 84.a ed. MacGraw-Hill; 1993. p. 2249-90.
- Zur diagnostik der tuberosen sklerose. Z Eerfosch Behandl. Jugendl. Schwachsinns, 1908;2:1-16.
- Eye sympthoms in phacomatoses. Trans Opthtal Soc UK 1932;52:380-401.
- Hamartial nature of tuberous sclerosis complex and its bearing on the tumor problem. Arch Intern Med 1942;69:589-98.
- Síndromes neurocutáneos discrómicos. Rev Neurol 1997;25(S):259-64.
- Facomatosis que cursan con manchas acrómicas, esclerosis tuberosa de Bourneville. Criterios diagnósticos y protocolos de seguimiento. Rev Neurol 1996;24(133):1056-9.
- Tuberous scleosis and allied disorders. Ann NY Acad Scienc 1991;615:397.
- Expressions of tuberous sclerosis complex gene products, hamartin and tuberin in central nervous system tissues. Acta Neuropathol 2000;99:223-
- Does tuberin function as a tumor supressor in the cerebral lesions of tuberous sclerosis? Early observartions. Brain Pathol 1996;6:375-6.
- Jancar J, Longuevidad de las personas con. sclerosis tuberosa. Medicina para el desarrollo y neurologia infantile 1996-Jul; 38 ( ) 652.
- Tuberous sclerosis. Arch Dermatol 1994;130:348-54.
- Curatolo P, Bombardieri R, Jozwiak S. (2008). Tuberous sclerosis. Lancet. 372: 657-668.
Pascual-Castroviejo I, Pascual-Pascual SI, Velázquez R, van den Ouweland AM, Halley DJ. (2006). Complejo esclerosis tuberosa tipo 1 (CET1): importancia diagnóstica de las minilesiones cutáneas en los casos de presentación familiar. Neurología. 21: 386-388. - Roach ES, Sparagana SP. (2004). Diagnosis of tuberous sclerosis. J Child Neurol. 19: 643-649.
Sparagana SP, Roach ES. (2000). Tuberous Sclerosis Complex. Curr Opin Neurol. 13: 115-119.
Grajkowska W, Kotulska K, Jurkiewicz E, Matyja E. (2010). Brain lesions in tuberous sclerosis complex. Review. Folia Neuropathol. 48: 139-149. - Staley BA, Vail EA, Thiele EA. (2011). Tuberous Sclerosis complex: Diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 127: 117-125.
- Discordant expression of tuberous sclerosis in monozigotic twins. J Dermatol 1991;18:178-80.
- Genomic imprinting: review and relevance for human diseases. Am J Hum Genet 1990;46:857-73.
- Tuberous sclerosis. Arch Dermatol 1995; 231:1460-2.
- Nódulos conjuntivales y dismorfia craneal en una esclerosis tuberosa atípica. Actas Dermosifioliogr 1989; 80:857-62.
- Esclerosis tuberosa de Pringle Bourneville. A propósito de 20 observaciones. Actas Dermosifiologr 1985;76:485-502.
- Esclerosis tuberosa. Med Clin 1991;96:784-7.
- The tuberous sclerosis syndrome: clínicasl and EEG studies in 100 children. J Neurol Neurosurg Psychiatry 1976;39:666-73.
- Cause of death in patients with tuberous sclerosis. Mayo Clin Proc 1991;66:792-6.
- Síndromes neurocutáneos con afectación ocular. Monogr Dermatol 1999; 12:181-90.
- B. Monteagudo Sánchez, C. de las Heras Sotos, E. León Muiños, J. Labandeira García, C. Durana Tonder, JMª Cacharrón Carreira. Placas de chagrín. An Pediatr 2007;66:421-2-Vol. 66 Núm.4 DOI: 10.1157/13101252.
- Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol 1996; 135:1-5.
- Roach ES, Sparagana SP. (2004). Diagnosis of tuberous sclerosis. J Child Neurol. 19: 643-649.
- Díaz T, Mateu A, Rojo R. (2010). Poliosis y status epiléptico en un lactante como presentación de esclerosis tuberosa. Actas Dermosifiliogr. 101: 812-814.
- Fitzpatrick T, Freedberg I, Eisen A, Wolff C, Austen F, Goldsmith L, et al. Dermatología en Medicina General. 5ta ed. Buenos Aires: Editorial Médica Panamericana; 2001. p. 110-7.
- www.orpha.net/data/patho/Han/Int/es/EsclerosisTuberosa_Es_es_HAN_ORPHA805.pdf
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous sclerosis complex: Genes, clinical features and therapeutics. Weinheim. Wiley-VCH. 2010. [ Links ]
- Baskin HJ Jr. The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol. 2008;38(9):936-52. [ Links ]
- Curatolo P, Maria BL. Tuberous sclerosis. Edited by PJ Vinken and GW Bruyn. Elsevier. Amsterdam. Handbook of Clinical Neurology 2013;111:323-31. [ Links ]
- Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics. 2008;9(7):475-87. [ Links ]
- Niida Y, Stemmer-Rachamimov AO, Logrip M, et al. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet. 2001; 69(3):493-503. [ Links ]
- Crino PB. The pathophysiology of tuberous sclerosis complex. Epilepsia 2010;51(Suppl 1):27-9.
- Meng XF, Yu JT, Song JH, Chi S, Tan L. Role of the mTOR signaling pathway in epilepsy. J Neurol Sci. 2013;332(1-2):4-15.