INFORMACIÓN BÁSICA.- Displasia Espondiloepifisaria Tardía (DEET). Tipo Kohn. (OMIM 271620).- La DEET tipo Kohn se caracteriza por enanismo de tronco corto, afectación progresiva de la columna vertebral y la epífisis y déficit intelectual de leve a moderado. El síndrome se describió en tres hijas nacidas de padres consanguíneos sanos. Las manifestaciones esqueléticas aparecen generalmente durante la infancia tardía. Las características radiográficas típicas incluyen: platispondilia, vértebras lumbares anormales y cambios degenerativos en las articulaciones grandes. Se ha sugerido una transmisión autosómica recesiva.
La DEE incluye un conjunto de trastornos cuya característica principal es el retraso en la osificación de las epífisis de huesos largos así como del esqueleto axial. Distinguimos varios trastornos dentro de este grupo de displasias óseas, como la acondrogénesis tipo II y la hipocondrogénesis, letales ambas en el recién nacido, y las formas congénita y tardía de la DEE propiamente dicha.
La DEE de comienzo tardío (DEET), exclusiva del sexo masculino, caracterizada por platiespondilia y afectación Epifisiaria limitada a las cabezas femorales. Cuando se agrega el déficit cognitivo se denomina tipo Kohn, trastorno presente en nuestro paciente.
Epidemiología.- La DEE confundida durante mucho tiempo con la enfermedad de Morquio, fue reconocida y descrita elegantemente como una entidad propia por Maroiteaux, Lamy y Bernard en 1975, en razón a sus peculiares componentes clínicos, su transmisión ligada al sexo y con afectación exclusiva en varones.
En 1973 según Berley, Nilsonne (1927) hace la primera descripción del cuadro clínico. El primer árbol genealógico ilustrativo y demostrativo fue publicado por Jacobsen 1939, sigue siendo la casuística más amplia, 20 miembros en 5 generaciones. Seis de estos pacientes fueron estudiados nuevamente 30 años después por Bannerman, confirmando el diagnostico de DEET.
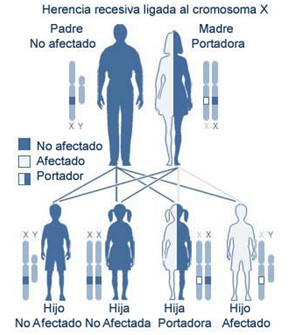
Herencia.- Autosómico dominante o Autosómico recesivo o Recesivo ligado al cromosoma X, estando por lo tanto afectados sólo los varones.
Genética.- La forma recesiva ligada al cromosoma X está causada por mutaciones en el gen TRAPPC2 (locus Xp22.2-p22.1).
Frecuencia.- La forma más común de esta enfermedad es la variante ligada al cromosoma X, con una prevalencia estimada de 1 entre 150.000-200.000.
En España, por dar un ejemplo del comportamiento de esta entidad, se ha contado con un caso dudoso, se han publicado 6 observaciones. La paciente de 67 años referida por Tagardo-Ferrero y cols., sin la característica alteración vertebral, probablemente padece de una displasia epifisiaria múltiple.
Recientemente en España el grupo de Miguel Cantalejo Moreira presenta un caso de un varón de edad media, con datos clínicos y radiológicos que sugieren DEET.
En nuestro país al carecer de estadísticas específicas, sería el primer caso publicado.
Etiopatogenia.- Con los métodos normales de investigación no se han detectado ningún trastorno metabólico ni déficit enzimático alguno (Lamy- Maroteaux, 1960). Esta afirmación sigue siendo válida a la actualidad.
En las formas clínicas atípicas, descritas por varios autores como O´Brien y cols., Byers y cols. Stanescu y cols., los hallazgos histológicos y bioquímicos hacen suponer mecanismos etiopatogénicos dispares.
Hallazgos tan dispares hacen suponer que están implicados mecanismos etiopatogénicos muy diferentes, pero capaces de originar parecidas alteraciones clínicas-radiológicas.
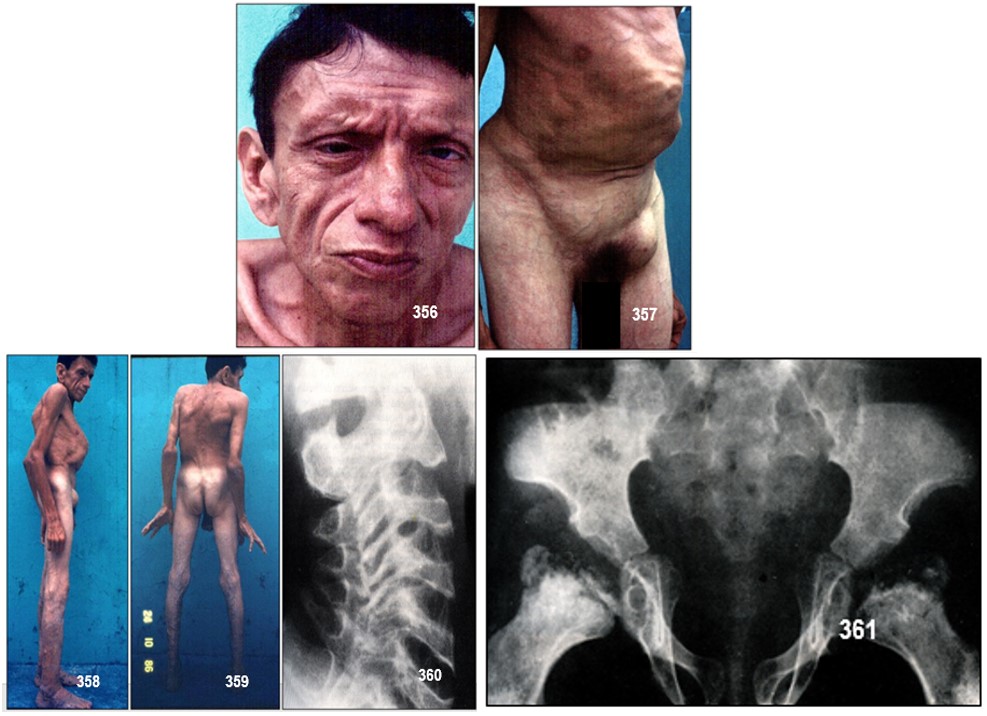
Manifestaciones clínicas.- Los pacientes pueden tener proporciones normales del cuerpo al nacer. Las manifestaciones clínicas no aparecen hasta la infancia, cuando se hace evidente la talla corta acompañada de dolor en caderas, rodillas y espalda. Los rasgos típicos de esta enfermedad son el tronco acortado, el pectus carinatum y la osteoartrosis prematura, particularmente en caderas y hombros.
Durante los primeros años de vida, el desarrollo somático suele ser normal. La enfermedad se manifiesta entre los 5 y años, en nuestro paciente se presentan a partir delos 2 años. A esta edad, el crecimiento del tronco parece detenerse, el tórax se ensancha progresivamente en sentido anteroposterior, los hombros van elevándose y el cuello se acorta.
Puede existir hipoplasia de odontoides o también os odontoideum, causas de inestabilidad atlantoaxoidea, la cual hay que descartar, y de escoliosis.
El adulto joven con DEET presenta una talla baja que oscila entre 127 y 160 cms, con una media de 140 cms, tronco corto, tórax ensanchado, protrusión esternal más o menos pronunciada y aumento de las incurvaciones fisiológicas de la columna vertebral.
En este momento, el estudio funcional de las articulaciones muestra una columna generalmente suficiente. La movilidad articular de los miembros es casi siempre normal, y cuando existe una limitación de los movimientos, suele ser discreta.
Radiología.- Las manifestaciones radiográficas aparecen antes de la pubertad y puede incluir: anomalías epifisiarias múltiples, cuerpos vertebrales aplanados, espacio discal estrecho, hipoplasia del proceso odontoides, cuello femoral corto y coxa vara.
Las alteraciones radiológicas de la DEET afecta casi exclusivamente a la columna, sin embargo, radiológicamente se aprecia afectación predominantemente de los hombros, caderas y rodillas, y platispondilia generalizada. Las imágenes radiológicas son diferentes en el niño y en el adulto.
La articulación coxofemoral está siempre afectada en coxa vara con aplanamiento leve de las epífisis, que conducen a cambios artrósicos precoces, y que pueden, en estadios más avanzados, incapacitar al paciente. A nivel axial se describen cambios displásicos vertebrales con platiespondilia, que provocan cifosis progresiva con aumento del diámetro anteroposterior del tórax, y que en último término puede provocar episodios de disnea. No aparece retraso mental, excepto en la forma clínica Tipo Kohn; se han descrito formas familiares con asociación a condrocalcinosis y existe una forma de displasia espondiloepifisiaria tarda asociada a artropatía degenerativa precoz, que puede simular artritis reumatoide juvenil.
Diagnóstico.- Es necesaria la asociación clínica y radiológica para establecer el diagnóstico de la enfermedad. El análisis genético molecular detecta una mutación en TRAPPC2 (SEDL) en más del 80% de los varones diagnosticados clínicamente.
Diagnóstico diferencial.- El diagnóstico diferencial de la DEET (no suele afectar al esqueleto axial) con la enfermedad de Scheuermann (la afectación vertebral es generalizada)1.
- – Displasia epifisiaria múltiple
- – Displasia espondiloepifisiria congénita; esta se manifiesta desde el nacimiento, la talla baja es más intensa y las alteraciones vertebrales son diferentes.
- – Enfermedad de Morquio
- – Ocronosis.
Tratamiento.- No hay tratamiento específico para la DEET. La sustitución protésica total, precedida o no de osteotomías correctoras, será necesaria en la mayoría de los pacientes a edades más tempranas que en la población general.
Posibles actuaciones quirúrgicas:
- Osteotomía correctora de coxa vara: Ángulo de inclinación del fémur mide unos 130 grados fisiológicamente; si este ángulo se cierra hablamos de coxa vara; si se abre hablamos de coxa valga. En cualquiera de los dos casos hay que hacer osteotomía (cortes) para abrirla o cerrarla respectivamente. Hacemos una varizante (en caso de coxa valga) o valguizante (en caso de coxa vara) según el caso.
- Endoprótesis en artrosis de cadera
- Fusión vertebral en hipoplasia odontoides
Bibliografía
- Elayne Esther Santana Hernández. Displasia espondiloepifisaria congénita. Revista de Ciencias Médicas de Pinar del Río, Vol. 22, Núm. 1 (2018).
- Arita M, Fertala J, Hou C, Kostas J, Steplewski A, Fertala A. Prospects and limitations of improving skeletal growth in a mouse model of spondyloepiphyseal dysplasia caused by R992C (p.R1192C) substitution in collagen II. PLoS One. 2017 Feb 9; [citado 18 Sep 2017] 12(2). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/28182776.
- Bayhan IA, Abousamra O, Rogers KJ, Bober MB, Miller F, Mackenzie WG. Valgus Hip Osteotomy in Children With Spondyloepiphyseal Dysplasia Congenita: Midterm Results. J Pediatr Orthop. 2017 Jan 30. [citado 18 Sep 2017].
- Singh SK, Rajoria K. Ayurvedic management of spondyloepiphyseal dysplasia tarda, a rare hereditary disorder. J Ayurveda Integr Med. 2016 Oct – Dec; [citado 11 Sep 2017]7(4):249-254.
- Saigal R, Chaudhary A, Pathak P, Singh A, Gupta D, Tank ML, et al. Spondyloepihyseal Dysplasia Congenita. J Assoc Physicians India. 2016 Jul; [citado 11 Sep 2017]64(7): 85-86.
- Srivastava P, Pandey H, Agarwal D, Mandal K, Phadke SR. Spondyloepiphyseal dysplasia Omani type: CHST3 mutation spectrum and phenotypes in three Indian families. Am J Med Genet A. 2017 Jan; [citado 18 Sep 2017] 173(1):163-168.
- Chung SW, Kang EH, Lee YJ, Ha YJ, Song YW. Three Cases of Spondyloepiphyseal Dysplasia Tarda in One Korean Family. Yonsei Med J. 2016 Sep; [citado 18 Sep 2017] 57(5):1290-3.
- Mitra S, Jindal S, Saroa R, Palta S. General anaesthesia for parturients with spondyloepiphyseal dysplasia: Risky but possible! Indian J Anaesth. 2016 Jun; [citado 18 Sep 2017]60(6):435-7.
- Nakashima Y, Sakamoto Y, Nishimura G, Ikegawa S, Iwamoto Y. A novel type II collagen gene mutation in a family with spondyloepiphyseal dysplasia and extensive intrafamilial phenotypic diversity. Hum Genome Var. 2016 May 19; [citado 18 Sep 2017]3:1600
- Liu L, Pang Q, Jiang Y, Li M, Wang O, Xia W. Novel COL2A1 mutations causing spondyloepiphyseal dysplasia congenita in three unrelated Chinese families. Eur Spine J. 2016 Sep; [citado 18 Sep 2017] 25(9):2967-74.
- Sangsin A, Srichomthong C, Pongpanich M, Suphapeetiporn K, Shotelersuk V. Whole-exome sequencing reveals a novel COL2A1 mutation in a patient with spondylo-epiphyseal dysplasia congenita. Genet Mol Res. 2016 Mar 11; [citado 18 Sep 2017] 15(1):15017624.
- Santolaya J.M, Degado A. Displasias Oseas. Salvat, 1988; 252-263.
- Bannerman RM, X-Linked spondylo-ephyseal displasia tarda. Birth Defects: Original Article Series, vol. IV. 48,1969.
- Cantalejo M, et al. Displasia espondiloepifisiaria tarda en varón de edad media. Reumatol Clin. 2011;7(4):267–268.
- Bleasel J, Bisagni-Faure A. Type II procollagen gene (COL2A1) mutation in exón 11 assocated with spondyloepiphyseal dysplasia, tall stature and precocious ostearthritis. J Rheumatol. 1995;22:255–61.
- Sillence DO. Displasias esqueléticas congénitas. En: Behrman RE, Vaughan VC, editors. Nelson. Tratado de Pediatría. Philadelfia: McGraw-Hill; 1989. p. 1469–85.
- Nilsonne, H. (1924): “Beitrag zur Kenntnis der kongenitalen Form der Coxa Vara”. Acta Radiol. 3: 383.
- Bleasel J, Bisagni-Faure A. Type II procollagen gene (COL2A1) mutation in exón 11 assocated with spondyloepiphyseal dysplasia, tall stature and precocious ostearthritis. J Rheumatol. 1995;22:255–61.
- Hedden F. Spondyloepiphyseal dysplasia. J Bone Joint Surg. 1978;60B:295.
- HamzaM, Bardin T. Camptodactyly, polyepipheseal and mixed crystal deposition disease. J Rheumatol. 1989;16:1153–8.
- Sambrook PN, de Jager JP, Champion GD. Synovial complications of spondiloephiphyseal dysplasia of late onset. Arthrtis Rheum. 1988;31:282–7.
- Bal S, Kocyigit H, Turan Y. Spondyloepiphyseal dysplasia tarda; four cases from two families. Rheumatol Int. 2009;29:699–702.
- Stanescu U, Stanescu R, y Maroteaux P. Pathogenic mechanisms in osteochondrodysplasias. J. Bone Joint Surg., 66A, 817, 1984.
- Tagardo-Ferrero D, Rosello Llerena J.A, y Rivera Redondo J. Displasia espondilo-epifisiaria tarda. Presentación de un caso de aparición tardía. Radiología, 29, 597, 1987.