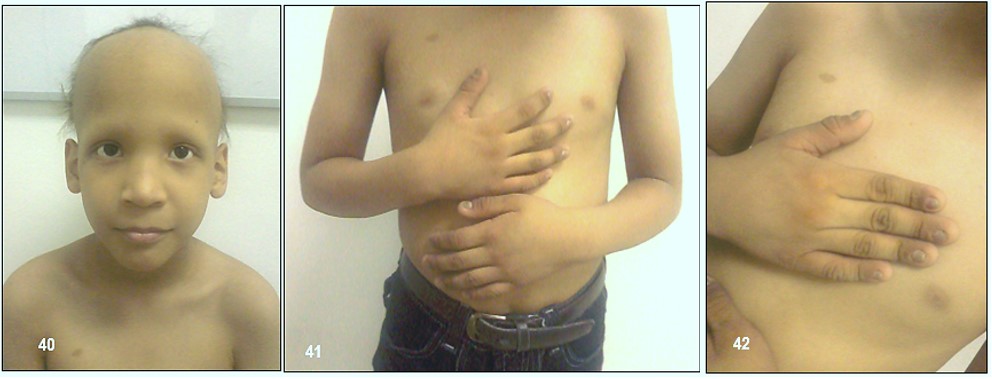
Imágenes 40-41-42.- Displasia Ectodérmica.- Paciente de 7 años de edad, sexo masculino, de una pareja de padres jóvenes y sanos, no consanguíneos, sin antecedentes familiares relacionados a esta enfermedad. Refería la madre que al nacer el niño presentó muy escaso cabello, de color claro y pajizo, ubicado únicamente en la región media posterior de la cabeza, con cejas y pestañas escasas, labios finos, uñas hipoplásicas y convexas, piel seca (hipohidrosis); puente nasal alto, e hipertelorismo ocular. Adviértase que todas estas manifestaciones se han mantenido exagerándose a tal punto que existe solo cabello de características finas (lanugo), en la parte coronal de la cabeza, ausencia total de cejas y uñas hipoplásicas, distroficas y quebradizas (Gentileza del Dr. Ramón Vargas Vera, Ecuador).
INFORMACIÓN BÁSICA.- Displasia Ectodérmica (OMIM:129490/ORPHA:189).- El término displasia ectodérmica (DE) engloba a un grupo heterogéneo de trastornos congénitos, los cuales comparten una alteración de la organización celular en al menos dos tejidos derivados de la capa embrionaria ectodérmica.
El ectodermo es uno de los tres componentes embrionarios primordiales, que alrededor de la tercera semana de desarrollo, experimenta una subdivisión en neuroectodermo, que dará origen al sistema nervioso, mientras que el ectodermo restante recubrirá toda la superficie embrionaria, y formará la epidermis, sus anexos y el esmalte dental.
Se estima una frecuencia de DE de 1 por 10.000 a 1 por 100.000 nacidos vivos, además se han descrito más de 170 subtipos clínicos diferentes. Las estructuras más comprometidas en la DE son las uñas, los folículos pilosos, las glándulas sudoríparas ecrinas y los dientes, aunque pueden existir manifestaciones como retardo mental, inmunodeficiencia y fisuras orolabiales. Tal variabilidad clínica ha llevado a proponer una nueva clasificación incorporando los conocimientos aportados por la genética molecular.
En términos generales, hay dos formas principales, clínica e histológicamente diferentes, la forma hidrótica o síndrome de Clouston y la hipo o anhidrótica o síndrome de Christ Siemens Touraine, según el grado de sudoración que presenta el paciente. Felsher sugirió el término de hipohidrótica ya que la piel raras veces es completamente anhidrótica.
Las formas hipohidróticas son reconocidas desde hace bastante tiempo. Una fue descrita por primera vez por Thurman en 1848, luego en 1875 el naturalista inglés Charles Darwin, durante un viaje a la India, reseñó una familia en que sólo varones estaban afectados, donde las mujeres eran sanas y transmitían la enfermedad a sus hijos, concordante con una herencia recesiva ligada al cromosoma X. Posteriormente, se han reconocido casos familiares con herencia autosómica dominante o recesiva. Clínicamente, sin embargo, estas formas son indistinguibles.
En la displasia ectodérmica hipohidrótica (DEH) la facies es típica desde los primeros meses de vida y se caracteriza por presentar: cejas, pestañas y cabellos escasos, gruesos y de color claro. Manifestaciones que se superponen a las de nuestro paciente. Se puede apreciar al nacer un patrón anormal de la implantación del cabello, con implante frontal alto (frente olímpica). Posteriormente, se describe calvicie prematura.
La piel es delicada, translúcida y lisa. Habitualmente hay áreas de hiperpigmentación y finas arrugas lineales alrededor de los ojos y la boca. Hay sequedad de piel y mucosas debido a una disminución del número y de la actividad de glándulas lacrimales, nasales, salivales y sudoríparas, siendo estas últimas las de mayor riesgo y relevancia. La falta de sudoración tiende a originar pirexia, en especial en climas cálidos o cuando los niños usan demasiada ropa. El ejercicio origina rubicundez o fatiga fácil. Desde lactantes puede observarse la falta de sudoración, pero no es raro que pase desapercibida durante años.
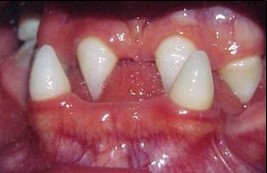
Imagen 6 con fines Didácticos.- DEH.- Ausencia de numerosas piezas dentales. Los dientes presentes muestran anomalías de forma, tamaño y estructura (dientes cónicos).
Estos pacientes suelen presentar un grado variable de hipoplasia medio facial, depresión del puente nasal, pómulos anchos, mandíbula triangular, labio superior corto y fino, labio inferior grueso y evertido, y orejas puntiagudas, pequeñas, de implantación baja y desplazadas hacia adelante. En la boca, disminuyen los rebordes alveolares, los dientes pueden ser cónicos puntiagudos, descoloridos y curvos, existiendo grados variables de hipodoncia e incluso anodoncia. La oclusión dentaria es pobre y las caries son comunes. La mucosa oral es seca, pudiéndose observar rinitis atrófica, disfagia, laringitis crónica y ronquera.
Las plantas y palmas son normales o rara vez hiperqueratósicas, las uñas de las manos se afectan en un 50%, siendo distróficas, estriadas, quebradizas y/o con convexidad acentuada.
Se puede acompañar con alteraciones inmunológicas como una susceptibilidad a infecciones, además de una dermatitis tipo atópica, rinitis alérgica y asma, con elevación de IgE sérica y alteración de la respuesta a pruebas de hipersensibilidad tardía.
Las glándulas mamarias pueden ser hipoplásicas o puede haber agenesia mamaria. El desarrollo mental de la mayoría de los pacientes es normal.
En la forma recesiva ligada al X, las portadoras suelen tener algunas manifestaciones que ayudan al diagnóstico de la forma de herencia, tales como: cabello frágil, ausencia de algunas piezas dentales o dientes cónicos, las glándulas sudoríparas pueden estar disminuidas y con una distribución anormal que sigue las líneas de Blaschko.
Aunque el diagnóstico es fundamentalmente clínico, con la triada de “3 H”, hipohidrosis, hipotricosis e hipodoncia, se puede complementar el estudio realizando una biopsia cutánea de la frente o de las palmas y plantas, la cual muestra disminución o ausencia de glándulas sudoríparas.
En la forma recesiva ligada al X, se puede realizar el test de sudor de Minor para identificar las portadoras que suelen tener algunas manifestaciones (cabello frágil, ausencia de algunas piezas dentales o dientes cónicos, glándulas sudoríparas disminuidas y con una distribución anormal que sigue las líneas de Blaschko. Consiste en impregnar la piel con yodo, agregando talco de almidón y luego generar calor (ejercicios o exponer a lámpara de calor) se evidenciarán puntos oscuros coloreados con yodo alrededor de la glándula sudorípara, siguiendo un perfil de distribución de las líneas de Blaschko.
Estos exámenes permiten detectar padres afectados con manifestaciones mínimas en las formas de herencia autosómica dominante, a excepción de los casos de mutación de novo o mutación germinal en cuyo caso los padres no estarán afectados. No consideramos necesario realizar dichos estudios complementarios ya que los padres no presentan manifestaciones.
Displasia Ectodérmica Hipohidrótica: Etiología / Genética molecular
La DEH es una enfermedad genéticamente heterogénea (con tres genes implicados hasta la fecha: EDA en Xq12-q13.1, EDAR en 2q11-q13 y EDARADD en 1q42.2-q43) y con tres patrones de herencia distintos: autosómica dominante (OMIM 129490), autosómica recesiva (0MIM 224900) y ligada a X (OMIM 305100).
Las formas recesiva ligada a X (secundaria a alteraciones alteraciones del gen EDA) y autosómica recesiva (secundaria a alteraciones en los genes EDAR y EDARADD) son clínicamente indistinguibles. La forma autosómica dominante (ligada también a los genes EDAR y EDARADD) presenta sintomatología mas leve.
Estos tres genes actúan en la vía de señalización de un factor nuclear, NF-kB, que interviene en la morfogénesis ectodérmica: EDA (OMIM 300451) activa EDAR (OMIM 604095) y utiliza EDARADD (OMIM 606603) como un adaptador para activar dicha vía. Se ha encontrado en pacientes con herencia autosómica recesiva, mutaciones en el gen de la proteína EDARADD (1q42), que actúa como moduladora intracitoplasmática del receptor EDAR. Estas proteínas son estructuralmente similares a otros componentes de la vía del factor de necrosis tumoral (TNF), y se expresan principalmente en la piel fetal, así como en el mesénquima subyacente. Se ha establecido que participan en la vía de señalización del factor de transcripción nuclear kappa B (NF-kB) como eje central (Imagen 7), que dirige la transcripción de genes necesarios para la formación de las estructuras epidérmicas.
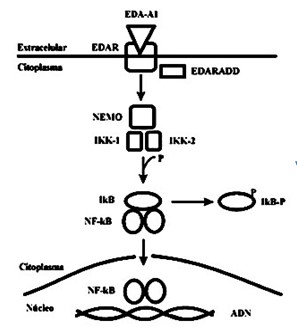
Imagen 7 con fines Didácticos.- Vía molecular del factor transcripcional NF-kB. La proteína EDAR es un receptor de transmembrana que se activa mediante la unión de su ligando (EDA-A1). La señal desencadenada es mediada por proteínas citoplasmáticas como EDARADD; y consiste en activar el complejo proteico IKK (compuesto por NEMO, IKK-1 e IKK-2) que a su vez fosforila a la proteína IkB. IkB normalmente está unido a NF-kB, reprimiendo su acción. Esta fosforilación provoca la liberación de NF-kB, que se traslada al núcleo para dirigir la transcripción de varios genes implicados en el desarrollo de estructuras ectodérmicas y otros tejidos. Estas mutaciones alteran la activación de NF-kB gatillada por EDAR, durante un período crítico en el desarrollo del ectodermo y sus apéndices, desencadenando la displasia. Esto ha sido corroborado en modelos animales. Se han encontrado mutaciones en otros componentes de esta vía, como el gen NEMO, que también es responsable de la Incontinencia Pigmenti.
En la forma recesiva ligada al X, las portadoras suelen tener algunas manifestaciones que ayudan al diagnóstico de la forma de herencia, tales como: cabello frágil, ausencia de algunas piezas dentales o dientes cónicos, las glándulas sudoríparas pueden estar disminuidas y con una distribución anormal que sigue las líneas de Blaschko.
El 95% de los pacientes con DEH seleccionados al azar presentan la forma ligada a X y el 5% restante la forma autosómica dominante (AD) o recesiva (AR).
El 95% de los pacientes con DEH seleccionados al azar presentan la forma ligada a X y el 5% restante la forma autosómica dominante (AD) o recesiva (AR).
A veces la historia familiar puede aclarar el modo de herencia y otras, especialmente si los casos son únicos en la familia, solo la identificación del defecto genético.
Concluyendo. A pesar de la baja incidencia de DEH, consideramos necesario que esta enfermedad pueda ser reconocida y sospechada por pediatras y neonatólogos, ya que su manejo debe ser multidisciplinario involucrando profesionales como pediatras, dermatólogos, genetistas, otorrinolaringólogos, dentistas, ortodoncistas, fonoaudiólogos, oftalmólogos y cirujanos plásticos. El diagnóstico precoz permite evitar el efecto deletéreo que puede acarrear la hipertermia y la susceptibilidad a infecciones respiratorias. Es necesario evitar y tratar la hipertermia aguda con control ambiental y baños o pulverizaciones de agua fría, que contribuirán a evaporar el calor corporal. Se debe informar al paciente y a la familia medidas para evitar el calor, ejercicio físico intenso, climas cálidos, se aconseja deportes acuáticos, deben evitarse medicamentos como clorpromazina, anticolinérgicos y diazepam.
La interacción de distintos genes y proteínas determina la heterogeneidad clínica en las distintas formas de herencia. Esto dificulta el diagnóstico de la enfermedad y del tipo de herencia, lo que puede llevar al clínico a dar información inexacta respecto al riesgo de recurrencia al paciente y sus familiares, interfiriendo con un consejo genético claro y preciso.
Cuando se presenta la DEH en las mujeres, la forma recesiva ligada al X es poco probable, debido a que las mujeres sólo son portadoras, con manifestaciones mínimas de la enfermedad. Sin embargo, no puede descartarse categóricamente esta herencia por el sólo hecho de ser mujer, ya que existen casos descritos de mujeres sintomáticas con DE ligada al X. Algunas de estas razones incluyen un sesgo en la inactivación del cromosoma X (por ejemplo, una alteración estructural que involucre al X) y la monosomía X, que pueden descartarse con el estudio cromosómico en sangre periférica.
Lo más factible es que corresponda a una forma autosómica recesiva donde ambos padres serían portadores sanos, y el riesgo de recurrencia en otro hijo sería de 25%, mientras que para la descendencia de la paciente, aún cuando todos sus hijos serían portadores obligados y clínicamente sanos, el riesgo de un hijo afectado sería prácticamente inexistente, ya que la frecuencia de portadores en la población general es muy reducida. Sin embargo, no es posible descartar que haya ocurrido una mutación de novo de una forma autosómica dominante, con lo que la probabilidad de recurrencia para los padres se haría muy remota, sin embargo, para los hijos de la paciente aumentaría a 50%. La precisión del patrón de herencia no es menor, no sólo por las probabilidades de recurrencia para hermanos de esta paciente, sino también para sus futuros hijos. El estudio molecular, que no está disponible en Ecuador, contribuiría a aclarar estas disyuntivas.
Bibliografía
- Gorlin RJ: Hypohidrotic ectodermal dysplasia. En: Syndromes of the Head and Neck. 3° Ed. Oxford Monografhs on Medical Genetics N°19, 1998; 451-5.
- Ruiz-Maldonado R, Parish LC, Beare JM, Paller AS: Displasia ectodérmica hipohidrótica En: Tratado de Dermatología Pediátrica 1° Ed. en español. México. Ed. Interamericana, S.A. 1992; 89-91.
- Kargul B, Alcan T, Kabalay U, Atasu M: Hypohidrotic ectodermal dysplasia: Dental, clínicasl, genetic and dermatoglyphic findings of three cases. J Clin Pediatr Dent; 26: 5-12.
- Zankl A, Addor MC, Cousin P, Gaide AC, Gudinchet F, Schorderet DF: Fatal outcome in a female monozygotic twin with X- linked hypohydrotic ectodermal dysplasia (XLHED) due to a de novo t (X;9) translocation with probable disrruption of the EDA gene. Eur J Pediatr 2001; 160: 296-9.
- Ellis SG, Alrmed H: Hypohydrotic ectodermal dysplasia affecting a female patient. Dent Update 1993; 20: 4450.
- Plottova-Puech I, Cambazard F: Dysplasies ectodermiques hypohidrotiques. Ann Dermatol Venereol 2002; 129: 1276-85.
- Priolo M, Lagana C: Ectodermal dysplasias: a new clínicasl-genetic classification. J Med Genet 2001; 38: 579-85.
- Sadler TW: Período Embrionario. En: Sadler TW. Langman de Embriología médica. 7° Ed. en español. Buenos Aires. Ed Médica Panamericana S.A. 1996; 62-83.