Históricamente, Say y Geraíd en 1968, describieron un Síndrome nuevo, en el cual 10 pacientes tuvieron múltiples malformaciones: además de ano imperforado presentaron polidactiíia, defectos vertebrales, estos casos fueron esporádicos y no había antecedentes familiares de defectos similares.
Say y colegas posteriormente ampliaron la asociación al incluir polioligodactilia. Quan y Smith utilizaron el acrónimo de AV para definir este conjunto de defectos que incluía: defectos vertebrales, atresia anal, fístula traqueoesofagica. displasía radial y renal.
La AV (a veces se lo designa como síndrome) designa un conjunto de anomalías, cada una de ellas producida por causas distintas, que afecta a varias estructuras del organismo y se debe a un trastorno producido entre la cuarta y la sexta semana del desarrollo embrionario. Es muy raro y sólo se han descrito unos 300 casos en todo el mundo. Por consiguiente es una enfermedad extremadamente rara del desarrollo embrionario, que afecta a diversas estructuras y vísceras del cuerpo humano.
El acrónimo VACTERL, también conocido como VATER o VATERS, procede de las iniciales de las estructuras embrionarias más implicadas en la enfermedad: vértebras, ano, corazón, región traqueoesofágica, riñón y extremidades inferiores, cuyo término anglosajón es limbs. En ocasiones, se presenta hidrocefalia, que extiende el acrónimo a VACTERL-H, Pero por otra para complicar aún más este síndrome, en algunos casos se presentan afecciones dentales y faciales por lo que algunos autores lo llaman VACTERL-DF.
Ya en el nacimiento o en los primeros días de vida se observa un grupo de malformaciones congénitas que incluyen al menos tres de las siguientes características: defectos vertebrales (60-80% de los pacientes), normalmente acompañados por anomalías de las costillas; ano imperforado/atresia anal (55-90%); defectos cardíacos (40-80%); fístula traqueo-esofágica (50-80%), con o sin atresia esofágica; anomalías renales (50-80%) incluyendo agenesia renal, riñón en herradura, y quistes y/o riñones displásicos; y anomalías en las extremidades (40-50%).
Los defectos en las extremidades se han definido clásicamente como anomalías radiales, incluyendo aplasia/ hipoplasia del pulgar, y tienen grados variables de gravedad; también se ha informado sobre otros tipos de anomalías en las extremidades. Mientras que las malformaciones arriba mencionadas son consideradas las características componentes básicas, se han descrito muchas otras malformaciones en los pacientes afectados. Estas malformaciones no típicas deberían ser usadas como claves a la hora de considerar otras posibles enfermedades.
El trastorno puede llegar a causar dismorfia en dos terceras partes de la zona inferior del cuerpo (tracto gastrointestinal distal, tracto genitoruinario, columna vertebral, pelvis y piernas) y a una tercera parte de la zona superior (atresia esofágica, anormalidades cardiacas y costales de tracto respiratorio). Desgraciadamente, el síndrome tiene mal pronóstico, causando una alta mortalidad en el primer año de vida (50-85 por ciento de los casos). Este hecho, sumado a que no se describió hasta los años 70, complica la investigación y el desarrollo de tratamientos, por lo que sólo se puede acudir a remedios paliativos. Se sabe que existen factores de riesgo, como la diabetes materna o la infertilidad, y se describe más en el sexo masculino que en el femenino (proporción de 2,6 a 1). La presencia de VACTERL en miembros de la misma familia hace pensar en trastornos genéticos (aunque no está demostrado), que quizá podrían estar implicados también en el origen de la hidrocefalia, dado que un mismo sujeto puede padecer ambas enfermedades.
Esta gama de hallazgos clínicos de presentación esporádicos son conceptualizados como asociaciones no aleatorias, que se presentan en al menos dos individuos, las cuales ocurren al mismo tiempo con más frecuencia que lo esperado al azar, sin ser componentes de síndromes conocidos.
Diagnóstico. – Actualmente no existe un consenso para el diagnóstico con respecto a cuáles o qué combinación de las regiones anatómicas descritas implicadas constituye un diagnóstico certero. No obstante, la existencia de una anomalía en cada una de las tres regiones anatómicas participantes (extremidades, tórax y pelvis, parte inferior del abdomen) permite su definición y diagnóstico.
Prevalencia. – Su prevalencia exacta y los datos de incidencia no están disponibles debido a los criterios de diagnóstico variables, pero se ha informado que la Asociación se produce en < 1-9/100.000 niños. Y la incidencia anual de 1:10.000 a 1:40.000 nacidos vivos. No se ha encontrado una distribución geográfica específica o un predominio en ciertos grupos étnicos.
Etiología. – Esta patología es incierta, se han planteado varias propuestas, entre ellas un defecto en la blastogénesis; en la diferenciación del mesodermo.
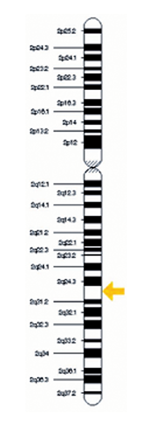 Imagen 3 con fines Didácticos:-
Imagen 3 con fines Didácticos:-
Esta condición resulta de eventos dismorfogenéticos que afectan la región cráneo-caudal del embrión. Los pacientes con el síndrome VACTER parecen formar un grupo distinto genéticamente y fenotípicamente, cuyo comportamiento en algunas familias es heredable de forma autosómica recesiva o ligada al cromosoma X. Otra sugiere una herencia de carácter autosómico sin evidencia de anomalías cromosómicas o teratógenicas, como en los casos de VACTERL con hidrocefalia (VACTERL-H) que siguen un patrón de herencia autosómico recesivo o ligado al cromosoma X; esta aparición es de carácter esporádico. Aunque se han encontrado algunos casos de Asociación VACTERL relacionados con trisomías y deleciones, generalmente no está vinculada con una anormalidad cromosómica.
Recientemente se han identificado microdeleciones del grupo de genes FOX en 16q24.1; cuatro genes, FOXF1, MTHFSD FOXC2, y FOXL1, se relacionan como causantes de un fenotipo similar a la Asociación VACTERL, que expresa anomalías vertebrales, atresias gastrointestinales (esófago, duodeno o ano), cardiopatías congénitas y malformaciones del tracto urinario, así como una rara anomalía letal de desarrollo del pulmón, la displasia capilar alveolar. La literatura reporta un caso donde se detectó una deleción intersticial 6q12 relacionada a la asociación VACTERL. Una mutación en el gen HOXD13 ha sido identificada en un paciente con muchas características de la asociación VACTERL. A manera de recordatorio, la proteína homeótica D13 es una proteína que en los humanos está codificada por el gen HOXD13.14 Este gen pertenece a la familia de genes homeobox. Los genes homeobox codifican una familia altamente conservada de los factores de transcripción que desempeñan un papel importante en la morfogénesis de todos los organismos multicelulares. Los mamíferos poseen cuatro grupos de genes similares homeobox, HOXA, HOXB, HOXC y HOXD, ubicados en diferentes cromosomas, que consta de 9 a 11 genes dispuestos en tándem. Este gen es uno de los varios genes homeobox HOXD ubicados en un grupo en el cromosoma 2 (véase la Imagen 2.). Las deleciones que eliminan todo el grupo de genes HoxD o el extremo 5 ‘de este grupo se han asociado con algunas anomalías genitales. Las mutaciones en este gen causan particularmente sinpolidactilia y braquidactilia. La participación de conocidos o previstos de la mutación del gen HOXD13 en las enfermedades humanas, el enlace NCBI Gene enumera las siguientes enfermedades o rasgos (fenotipos) se sabe o se cree que se asocia con cambios en el gen HOXD13:
- • Braquidactilia tipo D
- • Braquidactilia tipo E1
- • Síndrome Braquidactilia – sindactilia
- • La sindactilia tipo 5
- • Sinpolidactilia 1
Asociación VACTER El gen HOXD13 se encuentra en el largo (q) del brazo del cromosoma 2 en la posición 31,1 (flecha amarilla).
Más precisamente, el gen HOXD13 se encuentra desde el par de bases 176 957 531 176 960 665 al par de bases en el cromosoma 2. Por otro lado, la complejidad de la Asociación VACTERL con anomalías variables, como atresia anal, anormalidades en el rayo radial, genitales, así como alteraciones renales y cardíacas.
Existen otras malformaciones asociadas como son: defectos de los arcos branquiales, hipoplasia de páncreas, y otras que se siguen agregando haciéndola aún más compleja.
Debido a que existe una pluralidad clínica de casos VACTERL reportados a lo largo de la historia, en los que ninguno está ceñido a un patrón de presentación estricto, dando a conflictos nosológicos. Con el fin de evitar estos inconvenientes y con el objetivo de reconocer el fenotipo VACTERL en nuestro paciente, aplicamos los criterios de inclusión propuestos por Hall. De acuerdo a estos criterios una designación “segura” de VACTERL requiere al menos una anormalidad clave en cada una de las tres áreas topográficas del cuerpo involucradas (por ejemplo, miembros superiores, tórax y pelvis o abdomen inferior) mientras que una designación “probable” puede ser establecida por la ocurrencia de al menos 2 anomalías en dos de esas regiones.