INFORMACIÓN BÁSICA: Púrpura de Schönlein-Henoch (PSH).- La PSH es la vasculitis leucocitoclástica más frecuente en pediatría. Aunque pueda ocurrir a cualquier edad, se presenta más comúnmente entre los 5 y los 11 años, afectando más a varones que a mujeres. Las manifestaciones típicas de la enfermedad son: púrpura palpable, dolor abdominal (con o sin hemorragia gastrointestinal), artritis y compromiso renal (nefritis).
Etiología: Desconocida. Con frecuencia se encuentra el antecedente de afección del tracto respiratorio superior por el estreptococo betahemolítico del grupo A (10-30% de los casos), Yersinia, Mycoplasma pneumoniae, Bartonella henselae (más raramente) o por virus (EB, varicela, herpes simple, parvovirus B-19, etc.). Se ha identificado también infecciones micóticas, por protozoarios y por picaduras de insectos (abejas). Otros desencadenantes pueden ser fármacos (penicilina, ampicilina, eritromicina, quinina), alimentos, y exposición al frío.
Como factores genéticos predisponentes, se conoce que la PSH es más frecuente en niños con fiebre mediterránea familiar. Otros factores, como polimorfismos en el sistema renina-angiotensina, han sido implicados en el desarrollo de PSH y daño renal.
Patogenia: Es una vasculitis mediada por IgA de los pequeños vasos. Hay aumento en la producción de IgA, aumento de inmunocomplejos circulantes de IgA y depósitos de IgA en las biopsias de piel y de riñón. La lesión renal de la PSH es indistinguible histopatológicamente de la nefropatía por IgA de la enfermedad de Berger. Ambas pueden producir insuficiencia renal.
Manifestaciones clínicas: Las manifestaciones más importantes son cutáneas, articulares, gastrointestinales y renales.
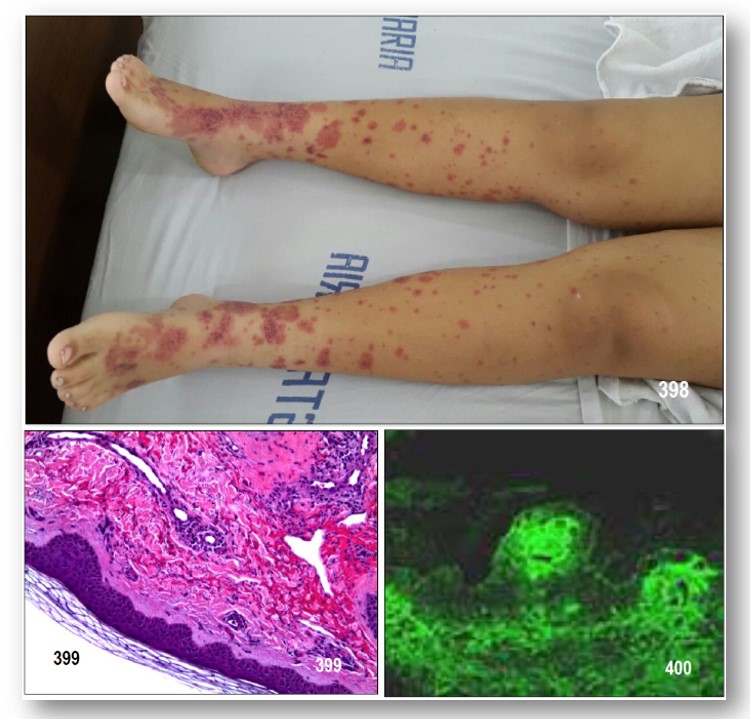
-Manifestaciones cutáneas. El exantema palpable (purpura palpable) eritematoso violáceo de tipo urticarial aparece en el 80-100% de los casos. Simétrico, en miembros inferiores y nalgas preferentemente, puede afectar cara, tronco y extremidades superiores. Regresa en una o dos semanas. Suele reproducirse al iniciar la deambulación. En niños menores de dos años se puede encontrar angioedema de cara, cuero cabelludo, dorso de manos y pies. En lactantes se ha llamado a este cuadro «edema agudo hemorrágico» o «vasculitis aguda leucocitoclástica benigna». Son cuadros eminentemente cutáneos con escasa participación renal o digestiva.
La púrpura palpable (erupción purpúrica, ligeramente elevada) tiene una distribución simétrica, con predilección por miembros inferiores y glúteos, pudiéndose observar también en miembros superiores (superficie de extensión de las articulaciones) y con menor frecuencia en tórax, abdomen, cabeza y cuello (Imagen 1). En casos severos pueden producirse lesiones equimóticas o necróticas. Algunos pacientes pueden presentar también edema subcutáneo doloroso, siendo característicos su aparición en cuero cabelludo. El dolor abdominal es de tipo cólico o difuso, periumbilical; se presenta en el 32 al 73% de los casos. A menudo empeora con la ingesta de alimentos, pudiendo estar acompañado de vómitos, hematemesis y melena.
-Manifestaciones articulares. El compromiso articular (artritis o artralgias) se presenta en más del 50% de los casos, afecta pocas articulaciones, más comúnmente grandes rodillas, tobillos). Estos pacientes tienen intenso dolor, sin proporción con los signos de inflamación. La artritis es transitoria, resolviéndose totalmente en pocos días, sin dejar deformidad permanente. La mayoría de los pacientes presentan desde el inicio lesiones en piel (rash), que incluso puede ser la primera manifestación en un 25% de los casos, y que pueden ser urticarianas o maculopapulares, apareciendo luego las lesiones purpúricas características. Esta erupción usualmente es acompañada por artralgias y dolor abdominal. Un pequeño grupo de niños puede presentar sólo dolor abdominal, apareciendo luego de varios días las les iones purpúricas.
-Manifestaciones gastrointestinales. El síntoma más frecuente es el dolor abdominal presente en el 40-85% de los casos. Se asocia con nauseas, vómitos si es grave, además, constipación o diarrea. Suele aparecer después del exantema, pero en un 14% de los casos puede preceder a los síntomas cutáneos, dificultando el diagnóstico.
Se puede encontrar sangrado en heces en la mitad de los casos (macro o micro).
El dolor abdominal se debe a la extravasación de sangre y líquidos dentro de la pared intestinal que puede llegar a ulcerarse, invaginarse (localización preferente ileo-ilial) o perforarse. También, aunque más infrecuentemente, puede haber una pancreatitis, un infarto intestinal o un hídrops vesical.
–Manifestaciones renales. Son las que marcan la gravedad o el peor pronóstico a largo plazo. Pueden ir desde una hematuria aislada microscópica hasta la presencia de una glomerulonefritis rápidamente progresiva. La prevalencia varía entre 20 y 50%.
La nefropatía se produce en la mayor parte de los casos en los tres primeros meses del comienzo de la enfermedad. Se suele asociar con afectación gastrointestinal y con la persistencia del exantema durante 2 o 3 meses.
El síntoma más común es la hematuria aislada. Más raramente se asocia con proteinuria. Si la afectación renal progresa, se produce un síndrome nefrítico con hematuria, hipertensión, azotemia y oliguria.
También puede aparecer un síndrome nefrótico con edemas y excreción de proteínas en orina de 24 horas > 50 mg/kg y cifras de albúmina en suero < 2,5 mg/dl.
Pacientes afectados de síndrome nefrítico y nefrótico conjuntamente desarrollarán fallo renal en un 50% en el plazo de 10 años.
La persistencia de proteinuria en rango nefrótico es predictiva de eventual fallo renal y debe ser revisada en Servicios de Nefrología con controles de biopsia renal. En ésta encontraremos patrones muy variados.
Por microscopia óptica se observa proliferación de células mesangiales, necrosis y proliferación extracapilar con aparición de medias lunas. Se puede clasificar la nefropatía de esta vasculitis en:
- Lesiones glomerulares mínimas.
- Progresión mesangial (focal o difusa).
- Formación de semilunas inferiores al 50%.
- Formación de semilunas entre el 50 y 75%.
- Formación de semilunas superior al 75%.
- Glomerulonefritis seudomesangiocapilar.
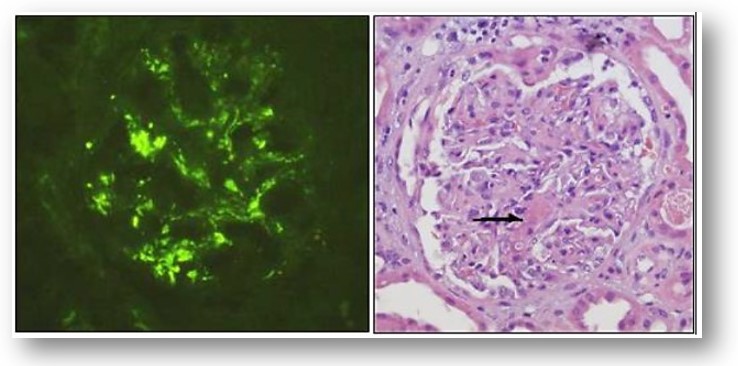
Por inmunofluorescencia y microscopia electrónica se observan depósitos de IgA en el mesangio del glomérulo, hallazgos que pueden ser similares a los de la nefropatía por IgA (NIgA), con mayor frecuencia de depósitos subendoteliales y subepiteliales, principalmente los primeros. En algunos casos los depósitos parietales predominan sobre los mesangiales. Al igual que en NIgA la subclase predominante en los depósitos es IgA. En las lesiones de piel pueden identificarse depósitos de IgA similares a los depósitos glomerulares.
Las alteraciones renales marcan el pronóstico de la enfermedad. Aproximadamente el 60% de pacientes presenta resolución de la enfermedad, usualmente en los primeros 6 meses; otro 15 a 20% presenta anormalidades urinarias menores sin deterioro de la función renal, y otro 20 a 25% presenta daño renal progresivo hasta falla renal terminal en 10 a 20 años.
Hallazgos de mal pronóstico incluyen un curso clínico prolongado, proteinuria severa, hematuria macroscópica, hipertensión arterial, porcentaje de semilunas (mayor del 50%) y, en algunos trabajos, extensión de los depósitos subendoteliales.
Otras manifestaciones clínicas menos frecuentes, son:
–Manifestaciones neurológicas. Cefaleas, cambios sutiles del comportamiento, hipertensión, hemorragias del SNC, y muy raramente neuropatías periféricas.
–Manifestaciones hematológicas. Diátesis hemorrágica, trombocitosis, déficit de factor VIII, déficit de vitamina K e hipotrombinemia que podrían producir una coagulopatía.
-Manifestaciones pulmonares. Neumonías intersticiales y, más graves, hemorragia pulmonar.
-Manifestaciones testiculares. Dolor, inflamación o hematoma escrotal con riesgo de torsión testicular.
Diagnóstico: Es clínico. No suele haber problemas en reconocer el cuadro si éste es completo, pero sí los hay si sólo domina un síntoma. Se han propuestos los siguientes criterios diagnósticos:
- Púrpura palpable. Lesiones cutáneas hemorrágicas «palpables» ligeramente elevadas, no relacionadas con Trombocitopenia.
- Edad de comienzo de la enfermedad menor o igual a 20 años. Paciente de 20 años o menor al comienzo de los primeros síntomas.
- Angina abdominal. Dolor difuso abdominal, que empeora tras la ingesta o el diagnóstico de isquemia intestinal, usualmente incluyendo diarrea hemorrágica.
- Granulocitos en biopsia. Cambios histológicos que muestran granulocitos en la pared de arteriolas y vénulas. Para decir que un paciente tiene Púrpura de Schönlein-Henoch, deben estar presentes al menos 2 de estos 4 criterios.
Diagnóstico diferencial: Con dolor abdominal. Invaginación u otro tipo de abdomen agudo quirúrgico. Con artritis. Fiebre reumática, poliarteritis nodosa, artritis reumatoide, LES.
- -Con exantema. Diátesis hemorrágica, reacción a fármacos, sepsis, malos tratos.
- -Con enfermedad renal. Glomerulonefritis aguda.
- -Con testículo doloroso. Hernia incarcerada, orquitis o torsión testicular.
•Examen clínico: Habrá que explorar detenidamente piel, presencia de edemas en cara, cuero cabelludo, escroto, etc., valorar las articulaciones con posible inflamación y palpación cuidadosa abdominal. 341 .
Exploraciones complementarias: No hay ninguna prueba diagnóstica selectiva. Puede haber moderada leucocitosis o eosinofília, anemia poco importante secundaria a sangrado, VSG y plaquetas elevadas si hay inflamación, aumento de las cifras de amilasa en caso de pancreatitis y diátesis hemorrágica secundaria a déficit del factor VIII. En orina se puede observar hematuria y ocasionalmente proteinuria.
Hay cifras elevadas de urea y creatinina en caso de fallo renal. Aquí estaría indicada una biopsia renal.
La prueba de sangre en heces con frecuencia es positiva.
Los niveles de IgA en sangre pueden ser normales o elevados. Aunque en el 50% de los pacientes esta elevada.
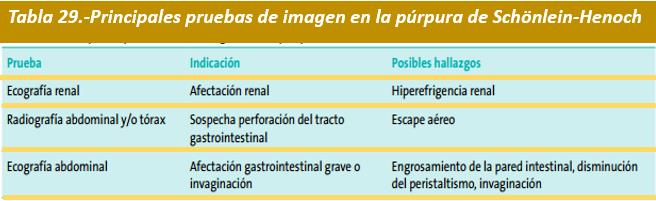
Pruebas de imagen.- De utilidad en pacientes con sospecha de complicaciones de la PSH. La Tabla 29 muestra las principales pruebas de imagen, sus indicaciones y posibles hallazgos. En caso de manifestaciones graves poco frecuentes, como afectación del SNC o pulmonar, pueden ser de utilidad otras exploraciones como la Radiografía de tórax, resonancia magnética cerebral, la angiografía o la tomografía computarizada cerebral. El enema baritado y la ECO abdominal deben realizarse en casos de dolores abdominales agudos intensos.
Evolución: Excelente la mayor parte de las veces. Autolimitada en 4 a 8 semanas. En casi la mitad de los casos tiene uno o dos brotes cada vez menos intensos. La hematuria aislada es el hallazgo de la mayoría de los casos con afectación renal. Sólo un 1% evoluciona a insuficiencia renal.
Tratamiento: No tiene. Se aconseja reposo en cama los primeros días. Los AINE se emplean para aliviar las molestias articulares.
Los corticoides están indicados a dosis de 1-2 mg/kg/día en casos de intenso de dolor abdominal o hemorragia intestinal y si aparece vasculitis en el SNC, afectación testicular o hemorragia pulmonar.
En los casos de deterioro renal, los esteroides suelen usarse u otros inmunosupresores, pero no hay acuerdo universal respecto al tratamiento óptimo. La enfermedad puede recurrir post-trasplante renal (53%), pero usualmente como depósitos de IgA sin deterioro de la función; se detecta nefritis clínica recurrente en aproximadamente 18% de casos; en 9% se producirá pérdida del injerto como consecuencia de la enfermedad.
Complicaciones: Urgencias (sospecha de invaginación, torsión testicular, etc.).
Cuando el cuadro está dominado por dolor abdominal, muchos de ellos son intervenidos quirúrgicamente con diagnóstico de abdomen agudo. La mayor incidencia de esta patología en primavera e invierno y el antecedente de infecciones del tracto respiratorio superior en gran número de estos pacientes, sugieren la presencia de un agente infeccioso que puede actuar como desencadenante de la enfermedad en una persona susceptible. Los organismos implicados son adenovirus, parvovirus, micoplasma, hepatitis B, varicela. La participación del estreptococo en esta vasculitis ha sido motivo de controversia, aunque actualmente sabemos que puede inducir el desarrollo de la enfermedad.
Seguimiento y pronóstico.- La PSH sin nefritis es una enfermedad autolimitada, con resolución completa de los síntomas en la mayoría de los pacientes. La duración es variable pero, generalmente, se resuelve en las primeras ocho semanas. Las recurrencias dentro del primer año afectan hasta un 30-40% de los pacientes y habitualmente son de menor intensidad y duración.
Una revisión sistemática comunicó que el 97% de los pacientes con daño renal lo desarrollaban en los primeros seis meses desde el debut. Por lo tanto, este es el mínimo tiempo de seguimiento recomendado, a pesar de que algunos autores alargan el seguimiento hasta los 12 meses. El riesgo de afectación renal en forma de hipertensión, proteinuria o disminución de la tasa de filtrado glomerular es del 2 al 15% según los estudios.
El riesgo de afectación renal en forma de hipertensión, proteinuria o disminución de la tasa de filtrado glomerular es del 2 al 15% según los estudios. El riesgo de insuficiencia renal terminal es <1%.
En los pacientes con afectación renal leve se recomienda un seguimiento a largo plazo anual para descartar la posibilidad de progresión de la afectación renal.
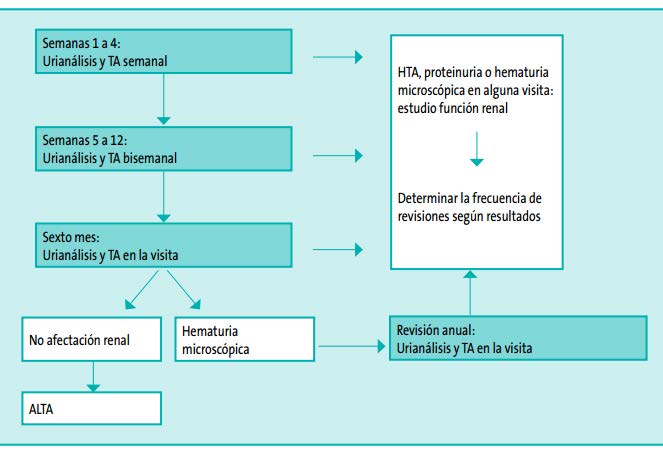
En la literatura se proponen diferentes protocolos de seguimiento para los pacientes con PSH para descartar afectación renal. Narchi et al. realizó una revisión sistemática de la literatura, comunicando que el 97% de los pacientes con daño renal lo desarrollan en los primeros seis meses desde el debut. Este mismo autor recomienda seguimientos periódicos mediante urianálisis y determinación de la tensión arterial (TA) al diagnóstico de la PSH y durante los primeros seis meses. Si todos los análisis son normales, el paciente no requiere seguimiento más allá del sexto mes. Si se detecta alguna alteración deberá estudiarse la función renal y realizar un seguimiento a largo plazo (urianálisis, TA y función renal) hasta la resolución de las alteraciones. Si se detecta un síndrome nefrótico o nefrítico, deberá valorarse urgentemente por un nefrólogo pediátrico.
La Figura 27 muestra una propuesta de seguimiento ambulatorio de los pacientes con PSH para descartar afectación renal (adaptado de McCarthy et al. y de Narchi et al.). En caso de episodios de PSH recidivante, se recomienda efectuar el seguimiento como en un primer episodio.
En las mujeres gestantes con antecedente de PSH en la infancia se recomienda un seguimiento cuidadoso de la gestación, ya que presentan una mayor incidencia de proteinuria y/o hipertensión durante el embarazo.
Bibliografía
- Bosch X, Font J, López-Soto A, Ingelmo M. Vasculitis. En: Enfermedades autoinmunes sistémicas y reumáticas. Barcelona, Doyma SA Ed. 1997: 121-136.
- Hills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purport. Arthritis Rheum 1990; 33: 1114-1121
- Jordi Vila Cots, jvilacots@hsjdbcn.org. Púrpura de Schönlein-Henoch, participación renal. An Pediatr Contin. 2012;10:121-6 – Vol. 10 Núm.3.
- Cassidy JT Petty RS. Vasculitis. En Cassidy JT, Petty RS, eds. Text book of Pediatric Rheumatology. 2nd. Ed. New York, Churchill Livingstone Inc; 1990: 377 – 423.
- Lanzkowsky S, Lanzkowsky L, Lanzkowsky P. Henoch-Schöenlein Purpura. Pediatr Rew 1992; 13: 130-137.
- Ballinger S. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2003;15:591-4.
- Cassidy J, Petty R, Laxer R, Lindsley C (eds.). Textbook of pediatric rheumatology, 6th ed. Philadelphia: Saunders Elsevier; 2011.
- McCarthy HJ, Tizard EJ. Clinical practice: Diagnosis and management of Henoch-Schönlein purpura. Eur J Pediatr. 2010;169:643-50.
- Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916-20.