INFORMACIÓN BÁSICA.- Osteogénesis Imperfecta (OI) (ORPHA:666/OMIM: 166200-166210-166220-166230-259420-259440 -610682/CIE-10:Q78.0): La OI fue un término difundido por Vrolik (1849) para designar este síndrome congénito de naturaleza genética, y de presentación variable, caracterizado por fragilidad ósea, osteoporosis y fracturas. Ekman en 1788 lo denominó osteomalacia congénita, Lobstein en 1853 osteopsatirosis, Eddowes en 1900 síndrome de las escleróticas azules, Porak-Durante distrofia periostal o síndrome de Van Der Hoeve (sordera en 1917).
Originalmente la enfermedad estaba dividida en dos tipos: Osteogénesis Imperfecta Congénita y Osteogénesis Imperfecta Tardía. Esta clasificación fue superada por investigaciones realizadas por el Doctor Sillence quién la dividió en cuatro tipos:
- Tipo I: es el tipo más frecuente, de transmite como autosomal dominante pero también puede ser el resultado de una mutación espontánea.
- Tipo II: abarca aproximadamente el 10% de las personas afectadas. Resulta de una nueva mutación y es la forma más severa que de Osteogénesis Imperfecta.
- Tipo III: abarca el 20%. Los enfermos sufren con frecuencia fracturas espontáneas.
- Tipo IV: es de leve a moderado. La mayoría de las fracturas se presentan durante la infancia.
Las clasificaciones no son siempre exactas hay pacientes que pueden compartir características de los tipos III/IV o II/III.
La osteoporosis, manifestación propia de trastornos hereditarios y adquiridos, se caracteriza clásicamente por fragilidad del sistema esquelético y predisposición para las fracturas de los huesos largos y las compresiones vertebrales causadas por traumatismos leves o insignificantes.
La OI (enfermedad de los huesos frágiles o de los hombres de cristal), causa más frecuente de osteoporosis hereditaria, es un trastorno generalizado del tejido conjuntivo debido a defectos del colágeno de tipo I. El espectro de la OI es sumamente amplio, y abarca desde una forma mortal en el periodo perinatal hasta una forma leve cuyo diagnóstico puede ser dudoso o ambiguo en el adulto.
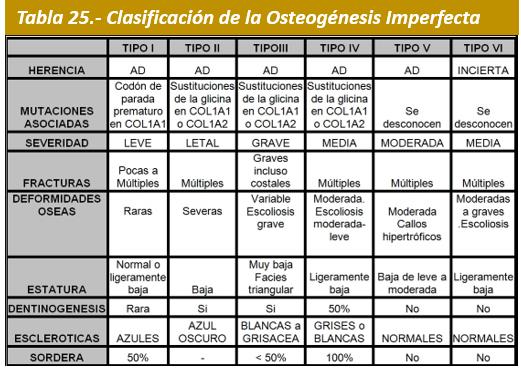
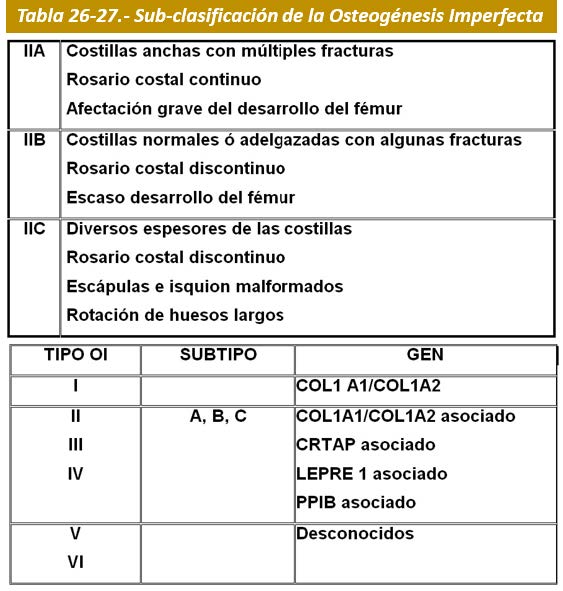
Clasificación.- La primitiva clasificación de Sillence (1979) dividía la OI en 4 tipos en base a criterios clínicos, radiográficos y genéticos, siendo el grupo IV el que presentaba mayor diversidad clínica. Aunque los criterios de Sillence fueron propuestos antes de que se identificasen los defectos del colágeno, todavía siguen siendo útiles cuando se adaptan a los nuevos conocimientos respecto a los defectos genéticos y a las distintas histomorfometrías. Sin embargo los pacientes no siempre encajaban en un grupo determinado de Sillence, por otra parte la histología ósea puso de manifiesto que pacientes con presentación clínica similar, podían presentar cambios muy diferentes en la organización del tejido óseo lo que llevo a Glorieux et al. a la definición de 2 tipos nuevos de OI (V y VI) (Tabla 25), basándose en sus distintas características clínicas e histológicas y que previamente se habían incluido en el grupo IV de Sillence. En 1984 se realizó una subclasificación del Tipo II en virtud de criterios radiológicos. (Tablas 26-27).
En 2006 Morello et al. describen el tipo VII producida por mutaciones en el gen CRTAP (proteína asociada al cartílago), cuya característica clínica es la rizomielia. En 2007 Cabral et al. describen el tipo VIII, asociada a mutaciones en el gen LEPRE1 , con fenotipo y sintomatología similares a los tipos II y III de Sillence (escleras blancas, severo retraso del crecimiento y extremada desmineralización del esqueleto).
En 2010, Aileen et al. describen el tipo IX en 2 casos de OI11, con herencia AR, debida a una mutación homocigota en el codón de inicio de PPIB, gen que codifica la CyPB (peptidil-propil cis-trans isomerasa). Clínicamente estos pacientes presentaban una OI de gravedad moderada, sin rizomielia, asociada a una hidroxilación normal de prolina 986 α1 y modificación normal de la hélice del colágeno.
En 2010, Van Dijk et al. efectúan una revisión de la clasificación de OI, y concluyen que si a cada nuevo gen que se va descubriendo, relacionado con la OI, se le asociase un nuevo tipo de OI, se crearía una clasificación ilimitada, basada en el gen afectado y no en las características clínicas del paciente, por lo que proponen una modificación de la clasificación, mencionando el gen causal y el cuadro clínico.
En 2011, Forlino et al. proponen una nueva clasificación (Tabla 28) que atribuye los originales 4 tipos de Sillence a mutaciones en COL1A1 y COL1A2. La OI tipo I debería limitarse a los casos con alteración cuantitativa del colágeno tipo 1, incluyendo aquellos individuos en los cuales la insuficiencia produce una clínica moderada.
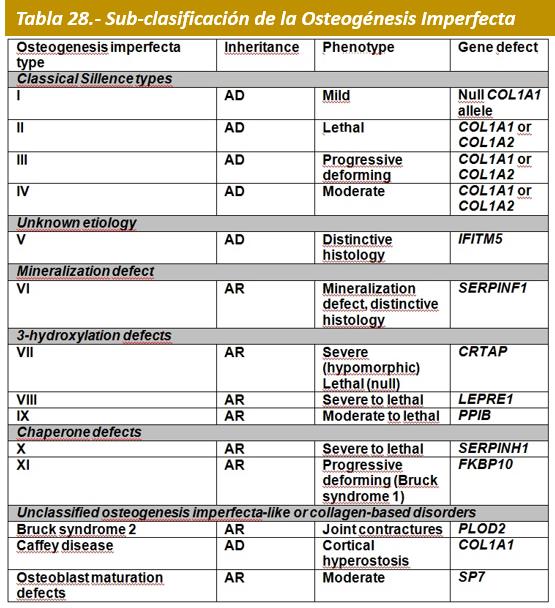
Los casos, en los que la mutación estructural del colágeno se asocia a un fenotipo muy leve deberían designarse como OI tipo IV. Esta clasificación asegura que la OI tipo I es un grupo homogéneo clínica y bioquímicamente, así como la única forma de OI dominante en la cual no hay colágeno anormal. Las formas recesivas de OI son diferenciadas también, según el gen en el que se produce la mutación o el producto genético afectado. Esta clasificación hace referencia de forma general al defecto genético y a la severidad del fenotipo, permitiendo generar grupos homogéneos para aproximaciones terapéuticas e investigaciones básicas del mecanismo de la enfermedad.
La clasificación de los pacientes, basada únicamente en criterios clínicos puede llevar a errores en el consejo genético, dado que pacientes con OI-AR y OI-AD pueden ser clínicamente semejantes, de ahí la importancia de la identificación del gen afectado.
En el 2012, Semler et al. han descubierto, como ya se ha comentado con anterioridad, que mutaciones del gen IFITM (se ha incluido en la tabla 4) son las responsables de la OI tipo V.
Características Clínicas .- Más adelante describo en detalles todas las manifestaciones clínicas
I/ Con herencia AD (tipos I al V)
En el tipo I, las fracturas con frecuencia aparecen con el inicio de la deambulación y disminuyen después de la pubertad.
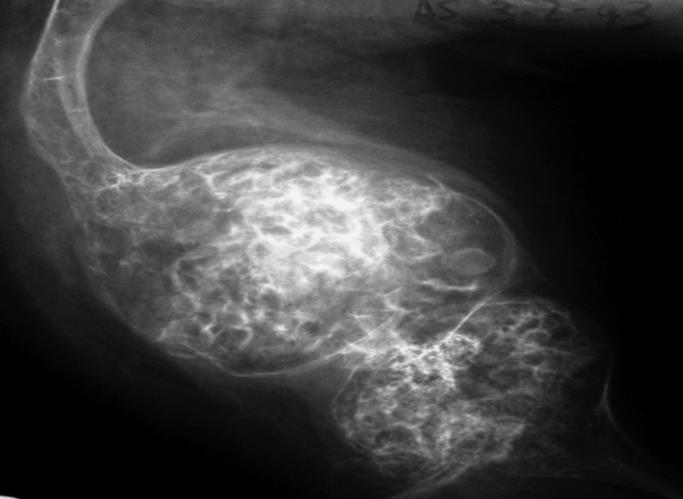
El tipo II es letal en el periodo neonatal, los lactantes afectos tienen huesos largos incurvados y cortos con múltiples fracturas intraútero, las escleras son azules o grisáceas, tienen un cráneo grande y débil. Las radiografías muestran huesos largos no bien tubulados. La causa más frecuente de muerte son fallos respiratorios asociados a un tórax pequeño con fracturas costales, neumonías y quizás alteraciones pulmonares a causa de anormalidades relacionadas con el colágeno.
La tipo III, progresivamente deformante, es la forma no letal más grave. Los afectados pueden tener hasta cientos de fracturas.
La mayoría tienen facies triangular con frente abombada, escleras azules o grisáceas, dentinogénesis imperfecta, compresiones vertebrales y escoliosis. Muchos tienen platibasia o impresión basilar. Tienen una talla muy baja y en la mitad de los casos tienen malformaciones en palomitas de maíz (Radiografía 1).
La tipo IV con afectación de moderada a grave, y un espectro clínico que se solapa con las formas I y III. OI
La tipo V, presentan como característica clínica que forman callos hipertróficos.
II/ Con herencia AR (tipos VI – XI) (Tabla 4)
a) Tipo VI, causada por mutaciones en el gen SERPINF1, que codifica el factor derivado del epitelio pigmentado o PEDF. En los pacientes con OI-VI y mutaciones truncantes de SERPINF1, el PEDF no se puede detectar en la sangre periférica, pudiéndose utilizar como parámetro de diagnóstico. Los experimentos con cultivos de células y en modelo in vivo proporcionaron pruebas de que el PEDF inhibe la diferenciación de los osteoclastos y, por lo tanto, la osteoclastogénesis a través de osteoprotegerina (OPG) y RANKL (el ligando de receptor activador para el factor nuclear k B). El activador del receptor de NF-kB (RANK), su ligando RANKL y el receptor señuelo OPG son reguladores centrales del desarrollo y la función de los osteoclastos. Clínicamente se parece a otras formas de OI (moderada a severa), sin embargo presenta hallazgos distintivos en la histología ósea, como un peculiar trastorno de las capas óseas (patrón de escama de pescado) y abundante osteoide no mineralizado, existiendo evidencias de un defecto grave de la mineralización.
b) Defectos en el complejo colágeno 3- hidroxilación
La propyl 3-hidroxilasa 1 (P3H1), la proteína asociada al cartílago (CRTAP) y el peptidil-propil cis- trans-isomerasa B (PPIB) se unen en un complejo 1.1.1 en el retículo endoplásmico, que postransducionalmente modifica los residuos específicos de prolina en las cadenas α del colágeno no plegado. Este complejo tiene también una función chaperona.
Tipo VII, se presenta con fenotipo moderado a letal, déficit de crecimiento, rizomielia, escleras blancas, severa osteoporosis, fracturas neonatales, huesos largos anchos y poco tubulados. Casi todas las mutaciones de CRTAP publicadas se corresponden a cuadros que resultan en NMD y una ausencia de la proteína CRTAP con pérdida también de la α1 3 hidroxilación.
Tipo VIII, presentan una enfermedad grave y en ocasiones mortal, con escleras blancas, rizomielia y disminución de la tubulación de los huesos largos. Aquellos que llegan a la infancia, tienen una densidad mineral ósea muy baja, así como déficit grave de crecimiento y metáfisis en forma de bulbo. Se deben a mutaciones en LEPRE1.
Tipo IX, se parecen a los tipos VII y VIII, pero no tienen rizomielia. Se debe a mutaciones en PPIB que producen un codón de parada prematuro o una alteración de la proteína.
c) Defectos de las chaperonas del colágeno
Los chaperonas son proteínas que ayudan al plegamiento / desplegamiento no covalente y ensamblaje / desensamblaje de otras estructuras macromoleculares. Las chaperonas no forman parte de la macromolécula cuando ésta realiza su función biológica, únicamente facilitan su ensamblaje o desensamblaje. La ausencia o disfunción de las chaperonas del colágeno SERPINH1 y FKB10 han sido descritos como los causantes de la OI tipos X y XI respectivamente.
Tipo X, el gen SERPINH1 codifica un chaperón de la molécula del colágeno llamado HSP47 (también conocida como proteína SERPINH1), que podría ser la responsable de monitorizar la parte final de la integración de la triple hélice del colágeno, se aloja en el retículo endoplásmico. Su función es la de actuar como un chaperón molecular específico de la molécula del colágeno (es el chaperón molecular del colágeno mejor conocido). Sin embargo el mecanismo molecular de su acción aún es controvertido; concretamente parece que HPS47 se une directamente a la triple hélice, a diferencia de los otros chaperones descritos que se unen a las cadenas alfa antes de formar la triple hélice.
El caso publicado con deficiencia de SERPINH1, describe a un niño con fenotipo grave, escleras azules y dentinogénesis imperfecta. Otros hallazgos presencia de bullas en la piel, estenosis de píloro y litiasis renal que requirió nefrectomía.
Tipo XI, el gen FKBP10, codifica un chaperón de la molécula del colágeno: la proteína inmunofilina FKBP65, localizada en el retículo endoplásmico. Alanay et al., describen 5 familias procedentes de la región norte de Turquía que padecían una forma severa y progresiva de OI. Se pudo establecer líneas comunes de descendencia en 2 de las familias, pero no en las otras 3; además de OI, estas familias padecían epidermolisis bullosa simple AR, resultante de un defecto en la keratina
Todos los individuos afectos presentaron: LRN y PRN normal, ampollas cutáneas al nacimiento en manos y pies, que con el tiempo evolucionaron a lesiones bullosas generalizadas, fracturas óseas recurrentes desde su infancia, osteopenia severa, deformidades en los huesos largos secundarias a las fracturas y cifoescoliosis con aplanamiento y acuñamiento vertebral. Ninguno de los pacientes presento dentinogenesis imperfecta ni escleras azules (escleras blanco-grisáceas) y su audición era normal. Algunos pacientes presentaron hiperlaxitud ligamentosa de los dedos de manos y pies, así como niveles elevados de fosfatasa alcalina (entre 300 y 400 UI).
El espectro fenotípico de las mutaciones en FKBP10 se solapan con el síndrome de Bruck tipo 1. Viljoen et al. describieron 5 niños con contracturas simétricas de rodillas, tobillos y pies, presencia de huesos wormianos y fracturas secundarias a traumatismos mínimos, dada la similitud con el caso publicado por Bruck en una revista médica alemana de 1897, propusieron denominar este trastorno Síndrome de Bruck. (Enfermedad AR caracterizada por osteoporosis, contracturas de las articulaciones al nacimiento, huesos frágiles y corta estatura), lo que a menudo es descrito como OI, con contracturas congénitas de las articulaciones.
d) Desórdenes del colágeno tipo 1 no clasificados
1) Sindrome de Bruck tipo 2, AR, causado por una mutación en PLOD2, que codifica para una hidroxilasa lisyl-telopéptido colágeno específica ósea (TLH). Los individuos afectos son clínicamente indistinguibles de aquellos con el S. de Bruck tipo1. La deficiencia de PLOD2 se traduce en una disminución de la hidroxilación de las lisinas del telopéptido de colágeno; pero no de la triple hélice lo que conlleva a un entrecruzamiento de las fibras de colágeno anormal.
2) Enfermedad de Caffey (Hiperostosis Cortical Infantil) AD, es un síndrome diferente, presentan sustituciones en el COL1A1 (p.Arg1014Cys) cuyos efectos sobre la matriz del colágeno causan síntomas de OI o de Síndrome de Enlers-Danlos.
3) Un defecto genético homocigoto en SP7 (factor de transcripción dedo de Zinc), también conocido como OSTERIX, se ha descrito en un niño con OI leve-moderada, disminución de la masa ósea vertebral, huesos wormianos, incurvación de los huesos largos, retraso en la dentición (no dentinogénesis), y deambulación, leve escoliosis, retraso de talla e hiperlaxitud de los dedos. Es un gen necesario para la diferenciación y maduración de los osteoblatos. La clasificación de este defecto, como causa de OI, es prematura en ausencia de datos bioquímicos, óseos y celulares dado que el SP7 no tiene un efecto directo selectivo en el colágeno tipo 1.
e) Nuevos genes asociados a OI – AR
Gen TMEM38B (un canal específico de cationes monovalentes involucrado en liberar Ca (2+) de los reservorios intracelulares), se encontro una mutación en 11 pacientes de los 27 estudiados en Arabia Saudi (11 familias y 2 aislados; pero con parentesco entre los padres) que tenian el estudio de COL1A1/A2 normal.
Gen BMP1/ mTLD, codifica la proteasa encargada de llevar a cabo el corte proteolítico del propéptido C-terminal del procolágeno I en el exterior celular, un proceso que es necesario para el correcto ensamblaje de las moléculas de colágeno en fibrillas y fibras. En 2012 mediante análisis de homocigosidad Martinez-Glez et al. encontraron una sustitución de la Fenilalanina 249 por Leucina en una familia egipcia con dos niños diagnosticados de una forma recesiva y grave de OI; los dos presentan numerosas fracturas, malformaciones óseas y hernias umbilicales. Asharani et al. identificaron la sustitución de la Glicina 12 por Arginina en una familia turca, este cambio altera la composición del péptido señal requerido para que esta proteína pueda ser secretada al exterior de la célula. Es interesante que mientras la mutación Gly12Arg parece estar asociada a huesos con masa ósea anormalmente elevada, los dos hermanos egipcios con la mutación Phe249Leu presentan un fenotipo aparentemente más grave y baja densidad ósea. Se podría pensar que la diferente posición de las mutaciones, localizadas en distintos dominios de la proteína podría explicar los diferentes fenotipos en lo relativo a densidad ósea, si bien podría simplemente tratarse de variaciones del fenotipo independientes del tipo de mutación, por lo que son necesarios más pacientes con mutaciones en este gen para poder llegar a una conclusión.
Gen WNT1, codifica un ligando de la ruta de Beta-catenina, una vía de señalización celular que desde hace tiempo se sabe es fundamental para la diferenciación y actividad de los osteoblastos. En 2013, varios investigadores Fahiminiya et al. y Keupp et al., han descrito mutaciones en el gen WNT1 (wingless-type MMTV integration site family, member 1) en familias con OI recesiva con múltiples fracturas y baja densidad ósea. Keupp et al. también describen mutaciones en este gen en una familia con osteoporosis dominante de aparición temprana.
Epidemiologia.- Abarca todos los grupos raciales y étnicos. Este trastorno óseo generalmente se presenta en el nacimiento como una enfermedad hereditaria. La Osteogénesis Imperfecta se clasifica en cuatro grandes tipos (y otros subtipos), todas ellas son ocasionadas por defectos en la cantidad o estructura del colágeno Tipo 1, el cual es una parte importante de la matriz del hueso. El problema con el colágeno usualmente resulta de un defecto genético dominante que puede ser adquirido por diversos y diferentes mecanismos:
- El defecto puede ser heredado en un patrón autosomico dominante de un padre afectado. Esto significa que un padre afectado que porta un gen único para este trastorno tiene un 50 % de posibilidades de tener hijos que lo padezcan y cualquier niño que lo herede resultará afectado.
- El defecto puede adquirirse por una mutación espontánea que se presenta en el óvulo o espermatozoide individual que formó al niño. En este caso, ninguno de los padres porta el gen para el trastorno o está afectado por el mismo. Los padres, en este caso, no tienen más riesgo que la población general para tener otro hijo con dicho problema.
- El defecto se puede adquirir a través de un patrón de herencia denominado mosaiquismo. Este fenómeno se presenta cuando un padre no está afectado, pero es portador de un porcentaje de espermatozoides u óvulos que portan el trastorno genético. Por lo tanto, aunque los padres no estén afectados, algunos de sus hijos
pueden tener el trastorno y otros no. Se estima que más o menos del 2 al 7% de las familias no afectadas que han tenido un hijo con osteogénesis imperfecta tendrán otro hijo con esta enfermedad debido al fenómeno de mosaiquismo.
Una de cada 20.000 personas padece Osteogénesis Imperfecta. Una de cada 50.000 a 60.000 personas desarrolla las formas más graves de la enfermedad.
Etiología.- Aunque existe una gran heterogeneidad genética, clínica y de pronóstico entre los diferentes tipos se admite que todas las formas tienen una similitud histológica y fisiopatológica debido a una delección de los genes Co1 1 A1 y 1 A 2, situados en 7q22.1 y 17q21.3-q22.5.
Todos los tipos de Osteogénesis Imperfecta se deben a defectos cualitativos o cuantitativos del colágeno de tipo I (principal componente de la matriz extracelular del hueso y la piel). Debido al alto predominio del colágeno en el hueso se produce una desmineralización ósea anormal, pero también a otros niveles: escleróticas, piel, dientes, oídos, etc.
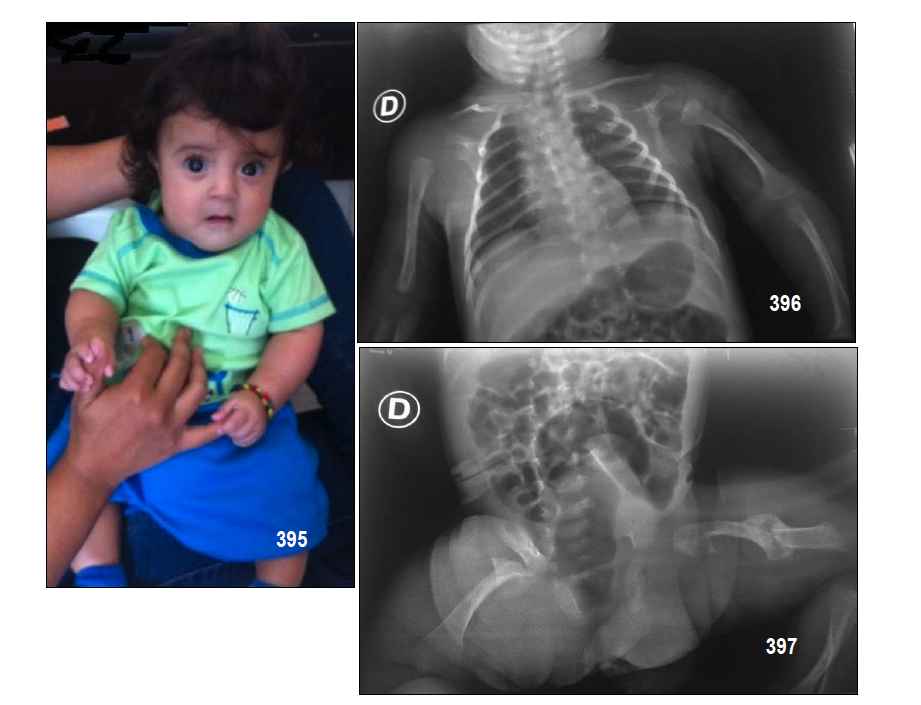
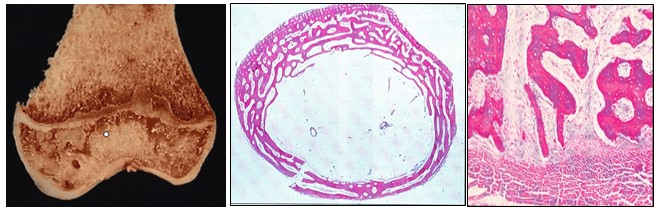
Anatomía patológica.- La matriz ósea contiene fibrillas anormales de colágeno tipo III y V. Los cristales de hidroxiapatita que se depositan en la matriz no están bien alineados con respecto al eje de las fibrillas. (Imagen 395).
Manifestaciones clínicas en detalles.– La compone una triada:
- Fragilidad ósea.
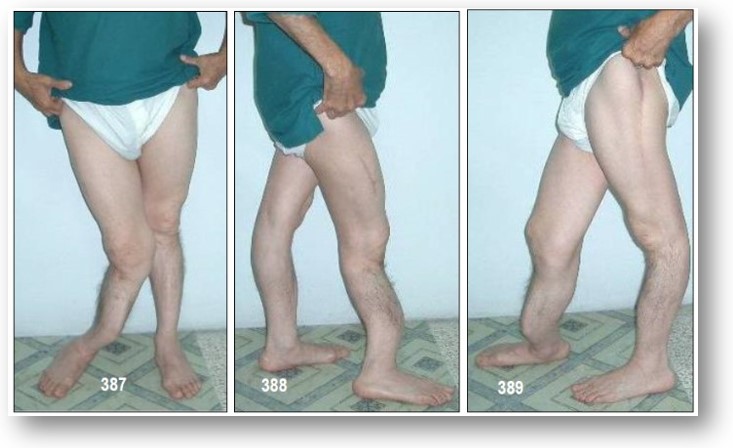
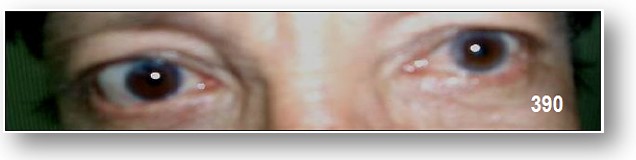
- Escleróticas azules. (Imágenes 390-395).
- Sordera prematura.
En los diversos tipos de Osteogénesis Imperfecta se puede encontrar una variedad de síntomas: –
- Fractura ósea:
- Presencia de más de un hueso fracturado en un sólo episodio (múltiple).
- Presente en el nacimiento.
- Después de un trauma menor.
- Deformidad de las extremidades o extremidades cortas.
- Sordera (la pérdida de la audición conductiva se puede presentar en adolescentes y adultos).
- Cifosis.
- Cifoescoliosis.
- Baja estatura.
- Deformidades dentales.
- Puente nasal bajo.
- Pectus carinatum (tórax en quilla).
- Pectus excavatum (tórax excavado).
- Pes planus (pie plano).
- Laxitud de las articulaciones.
- Hipermovilidad.
- Tendencia a la formación de hematomas.
- Piernas en arco.
- Voz aguda.
- Estreñimiento.
- Sudoración excesiva.
- Músculos débiles.
- Rostro en forma triangular.
- Huesos Vormianos (pequeños osículos dentro de las líneas de sutura craneana, percibibles en las radiografías del cráneo).
Sordera
La pérdida significativa del sentido del oído se da aproximadamente en el 50% de las personas con Osteogénesis Imperfecta. En el caso de Osteogénesis Imperfecta tipo I, la forma más frecuente de aparición de pérdida del oído, comienza alrededor del los 20 – 30 años.
Hay tres tipos principales de alteración:
- Conducción: resultado de un problema físico en el oído medio o externo; puede ser consecuencia de una infección del oído, obstrucción o por fractura de la cadena de huesecillos que se encuentran en el oído, por dentro del yunque.
- Neurosensorial: cuando el oído interno no transmite la señal de sonido de forma adecuada al cerebro.
- Mezcla: cuando están implicados el oído medio e interno.
La pérdida del oído también se puede clasificar según el grado de severidad (leve, moderada, severa y profunda) o por la frecuencia afectada (bajo, alto o todas las frecuencias).
Los síntomas que podemos encontrar son:
- Dificultad para entender ciertas palabras o partes de palabras.
- Preguntas frecuentes al interlocutor para que éste repita las palabras.
- Dificultad para entender por teléfono.
- Volumen excesivo de la televisión o la radio.
- Sensación de entorno ruidoso.
Si el mal progresa, la pérdida del sentido del oído puede interferir con la comunicación normal, el rendimiento en el trabajo y en las actividades sociales y personales. Si no se trata adecuadamente puede provocar aislamiento y depresión.
-OSTEOGÉNESIS IMPERFECTA TIPO I (LEVE)
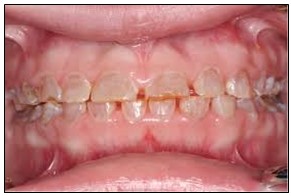
También llamada enfermedad de Lobstein. Presenta la tríada clínica en el 30-60% de los casos. A su vez la tipo I se subdivide en A o B según falte o exista dentinogénesis imperfecta, dientes descoloridos y frágiles debidos a la defectuosa formación de la dentina, que conlleva la decoloración y la fragilidad de los dientes (Figura 4).
Herencia: Autosómica dominante, la frecuencia de este tipo de osteogénesis imperfecta es de 1 en 15 mil a 1 en 20 mil nacidos vivos, pero por su presentación clínica leve puede ser más frecuente.
Etiopatogenia: Disminución en la producción de proalfa 1, substitución por otros residuos distintos a la glicina en la triple hélice de alfa 1.La disminución de la producción de proalfa 1 no es la única vía de disminución de la secreción de procolágena sin secretar moléculas anormales, por ejemplo, la síntesis de proalfa 2, cadena que pudiera ser incorporada dentro de las moléculas normalmente, pero que presentan una rápida y completa degradación intracelular teniendo así un efecto similar.
Cuadro Clínico: Los defectos que se mencionaran a continuación se manifiestan aproximadamente entre 25 y 60 % de los pacientes con osteogénesis imperfecta tipo I. Se ha sugerido que este grupo de pacientes pudiera subdividirse en base a la ausencia tipo IA o presencia tipo IB de dentinogénesis imperfecta.
Los individuos afectados tienen peso y talla al nacer normal, rara vez tienen fracturas en el periodo perinatal, aunque el arqueo femoral intrauterino y fracturas al nacimiento pueden ser la presentación inicial.
Las fracturas pueden ocurrir primero dentro de las semanas posteriores al nacimiento asociadas al realizar el cambio de pañal pero más comúnmente ocurren cuando los niños comienzan a caminar. Los huesos que más frecuentemente se rompen son los huesos largos de brazos y piernas, las costillas y los huesos pequeños de manos y pies. La frecuencia de las fracturas permanece constante a través de la niñez y disminuye posterior al inicio de la pubertad, sugiriendo que los factores hormonales y otros alteran la fuerza ósea. Las fracturas sanan rápidamente con evidencia de buena formación de callo y sin deformidad. La osteoporosis es marcada y la producción de escasa deformidad ósea. La morfología de los cuerpos vertebrales es normal en un inicio, pero con frecuencia se desarrolla la clásica apariencia de bacalao que se acompaña de pérdida de talla en las décadas posteriores.
La facies es proporcionalmente pequeña, triangular, el cráneo es delgado y blando, con fontanelas amplias y huesos wormianos. Las regiones temporal y frontal son protuberantes con platibasis en la base del cráneo. Las articulaciones y ligamentos son hiperextensibles, lo que lleva algunas veces a cifoescoliosis, pies planos y en casos extremos a luxación articular.
En la dentición hay hiperplasia de dentina y pulpa con translucidez de los dientes, los cuales tienen coloración amarilla o gris azulada. Existe susceptibilidad a las caries y colocación irregular, así como erupción tardía de los dientes.
La piel y las escleróticas tienden a ser delgadas y translúcidas. La coroides es especialmente visible y da a las escleróticas apariencia azulada.
Otras alteraciones son hernia inguinal y/o umbilical, escaso desarrollo muscular, una mayor tendencia a la hemorragia secundaria, a la fragilidad capilar y/o a la función plaquetaria anormal. Dentro de las anormalidades ocasionales se encuentra a nivel de ojos el embrotoxon (opacidad periférica de la córnea), queratocono y megalocórnea. Otros son sindactilia y válvula mitral flácida.
Cerca de la mitad de los individuos afectados tienen pérdida de la audición de inicio temprano, la cual es típicamente de alta frecuencia y la sordera es de conducción ya que el trastorno del colágeno afecta a la cadena de huesecillos del oído medio. De los 30-39 años de edad el 35% de los pacientes es sordo y el 50% presenta sordera a los 60 años de edad.
Diagnóstico: Es sospechado clínicamente por la típica triada de Van der Hoeve: fragilidad ósea, escleróticas azules y sordera, además de tener una historia familiar dominante. El diagnóstico se confirma al medir la producción de procolágena tipo I por fibroblastos dérmicos en cultivo.
Normalmente, cerca de 85% del colágeno sintetizado por los fibroblastos es procolágeno tipo I y la mayor parte del resto es procolágeno tipo III, las células de los pacientes con osteogénesis imperfecta tipo I sintetizan cerca de la mitad de la cantidad normal de procolágeno tipo I pero una cantidad normal de otras proteínas.
El diagnóstico es particularmente importante en los niños afectados en los que no hay historia familiar ya que esto facilita el consejo genético y ayuda a concientizar a la familia acerca del pronóstico.
Diagnóstico Diferencial: Hiperlaxitud e hipotonía (conectivopatías congénitas), enfermedad de Ehlers Danlos, fracturas, displasia ósea, fragilidad ósea.
Diagnóstico Prenatal: La identificación prenatal de fetos afectados puede permitir por algún método identificar el alelo mutante, los cuales incluyen estudios de segregación con una estructura familiar apropiada, identificación de los alelos no expresables en el RNA mensajero del feto o al reconocer fracturas o arqueo de los huesos largos por ultrasonido. Los estudios de segregación de alelos pueden proveer el diagnóstico a las 11-12 semanas de gestación si se utilizan las muestras de vellosidades coriónicas. El análisis de la cantidad de procolágeno tipo I sintetizado por células cultivadas de muestras de vellosidades coriónicas tomadas en la novena-décima semana de gestación puede proveer un medio para detectar fetos afectados tempranamente en la gestación.
Evolución natural: Existe una amplia variabilidad individual con mortalidad temprana de lactantes severamente afectados, relacionada principalmente con bronconeumonía. Después de la lactancia la perspectiva de sobrevivencia es buena. La deformidad de miembros aparece en 20%, lo mismo que la cifoescoliosis, la necesidad de férulas o prótesis en 1 de cada 10 casos, silla de ruedas excepcional.
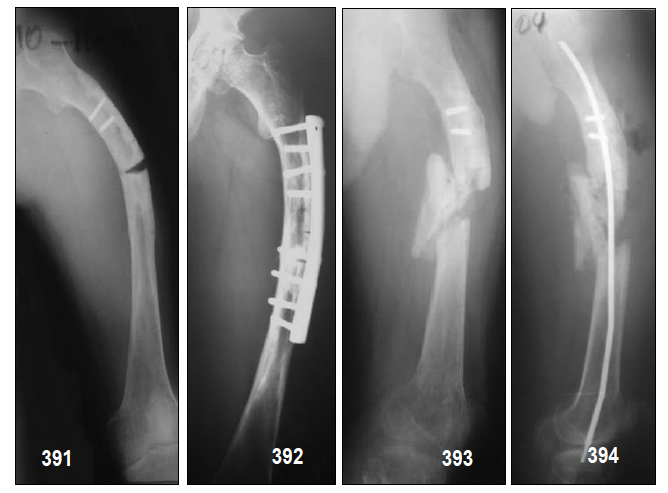
Tratamiento: Un soporte intramedular mediante varillas metálicas debe ser considerado como tratamiento de los casos graves con fracturas múltiples y un óptimo manejo ortopédico. Recientemente, ha sido comprobado que la calcitonina administrada a largo plazo, puede favorecer la disminución en la frecuencia de fracturas. También, el fluoruro, al producir un cristal más fuerte de fosfato de calcio, puede ser benéfico.
Con la producción de hormonas sexuales en la adolescencia generalmente hay una mejoría parcial.
-OSTEOGÉNESIS IMPERFECTA TIPO II (LETAL)
También llamada enfermedad de Vrolik.
Herencia: autosómica dominante (nueva mutación). Autosómica recesiva (rara). Se afecta entre 1 a 20 mil y 1 en 60 mil infantes.
Etiopatogenia: Reagrupamientos en los genes COL 1ª1 y COL 1ª2, substituciones por residuos de glicil en la triple hélice de alfa 1 (I) o alfa 2 (I),
Cuadro Clínico: Prematurez y bajo peso al nacer son comunes. Los infantes afectados tienen una facies característica, con escleras azul intenso, nariz pequeña y con puente nasal bajo (pico de águila), la bóveda craneana es extremadamente suave, escasamente mineralizada con fontanelas grandes y múltiples huesos wormianos, macrocefalia relativa. Las extremidades son cortas, gruesas, las piernas arqueadas y las caderas están usualmente en flexión y abducción, la cavidad torácica es generalmente muy pequeña. Se observan múltiples fracturas y callo óseo desarrollado en especial en miembros inferiores. Otras alteraciones son vértebras aplanadas, hipotonía, hernias inguinales, hidrocefalia variable.
Sillence et al., basados en diferencias observadas en las radiografías, subdividieron recientemente este trastorno en 3 grupos .Grupo A se caracteriza por fémures encogidos, anchos y cortos y costillas como ”cuentas de rosario”, grupo B presenta fémures encogidos, anchos y cortos con costillas normales o como cuentas de rosario incompletas. Grupo C se distingue por huesos largos rectangulares, inadecuadamente moldeados, delgados y largos con múltiples fracturas y costillas delgadas y arrosariadas.
La muerte usualmente resulta por falla respiratoria y frecuentemente ocurre durante las primeras horas posteriores al nacimiento. Más de 60% de los recién nacidos mueren durante el primer día de vida, 80% mueren en el primer mes y la sobrevivencia más allá de un año es rara.
Diagnóstico: Con el incremento en el uso rutinario tempranamente del ultrasonido gestacional, los primeros niños afectados en las familias son detectados durante el segundo trimestre del embarazo. Las características tempranas en el ultrasonido durante el embarazo pueden ser difíciles de distinguir de otras formas letales de displasia esquelética.
Las características anormalidades de las costillas y marcada disminución en la mineralización de la bóveda ósea son las características más útiles. Si hay alguna duda, el diagnóstico de osteogénesis imperfecta tipo II puede ser confirmado por examen de las fibras de colágena sintetizadas por fibroblastos cultivados en uno o varios tejidos. Alternativamente, si no hay fibroblastos disponibles para estudiar, el examen patológico con el descubrimiento de hueso desmineralizado, células osteoblásticas dilatadas e incremento osteoide puede ayudar a confirmar el diagnóstico.
Diagnóstico Diferencial: Displasias esqueléticas letales especialmente la displasia tanatofórica y acondrogénesis, así como la hipofosfatemia autosómica recesiva
Diagnóstico Prenatal: Ha sido completado por la utilización del ultrasonido obstétrico a embarazadas entre las semanas 14 a 18 de gestación. Para esa edad los fémures son cortos, la caja torácica es pequeña y la mineralización de la bóveda craneana es mínima. Análisis de las vellosidades coriónicas por la presencia de fibras de colágena anormales y sintetizadas por células creciendo de vellosidades coriónicas pueden ser utilizados para excluir el diagnóstico y para identificar un feto afectado.
-OSTEOGÉNESIS IMPERFECTA TIPO III (DEFORMANTE PROGRESIVA)
La variedad deformante progresiva es reconocida usualmente al nacer por estatura corta y deformidades resultantes de fracturas útero.
Herencia: Hay forma autosómica dominante y recesiva las cuales han sido bien documentadas, aunque la forma recesiva es raraen la mayoría de las poblaciones
Etiopatogenia: En los casos autosómicos dominantes hay una mutación puntal en la cadena alfa 1 o alfa 2. En los casos autosómico recesivos existe una mutación que previene la incorporación de proalfa 2 dentro de las moléculas.
Cuadro Clínico: Este tipo de osteogénesis es una variedad progresiva deformante, la cual usualmente es reconocida al nacimiento por estatura corta y deformidades resultantes por fracturas in útero. Por la fragilidad ósea y deformidad, muchos de estos niños desarrollan cifoescoliosis significativa y puede progresar a insuficiencia respiratoria. Radiológicamente al nacimiento la bóveda craneana presenta pobre mineralización, las costillas son delgadas, los huesos largos son delgados con evidencia de fractura y el esqueleto es osteopénico. Si las fracturas no están presentes al nacimiento, éstas ocurren usualmente durante el primer año de vida y la deformidad llega a ser aparente en este periodo. Estas se observan entre los 2 a 5 años de edad, estructuras quísticas inusuales en las epífisis de algunos huesos largos, especialmente en fémur pueden ser encontradas. Esto da como resultado que el crecimiento óseo sea pobre afectándose la talla. Por la corteza delgada de los huesos, la fractura de los mismos es frecuente. La deformidad en los ángulos de la tibia y fémur reduce la eficiencia de los mismos e incrementa la posibilidad de fractura.
Incluso dentro de OI tipo III, se observa una gran heterogeneidad a nivel clínico .La distinción entre un individuo con el tipo leve III OI y OI tipo IV es a menudo sutil, y muchos médicos utilizan la capacidad para caminar de forma independiente para diferenciar los dos. La resistencia mecánica del hueso como se determina por indentación nano es la misma en ambos subtipos y independiente del tipo de hueso.
Algunos individuos tienen dientes de aspecto normal y la apariencia facial, mientras que otros tienen DI y relativa macrocefalia con prominencia frontal. Ventrículos agrandados que reflejan la bóveda craneal suave y un tercio medio facial aplanado se observan con frecuencia. Barril deformidad torácica y cifoescoliosis también son comunes. Tonalidad escleral es variable, a menudo de color azul o gris, y la esclerótica puede ser de color azul en la infancia, pero aligerar con la edad. La pérdida de audición por lo general comienza en la adolescencia.
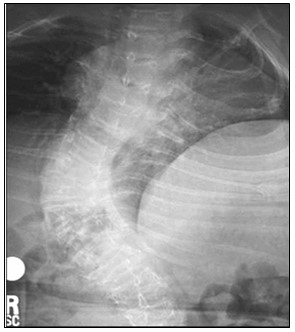
La Impresión basilar, una anormalidad de la unión craniovertebral causada por el descenso del cráneo sobre la columna cervical, es una complicación conocida, la forma sintomática ocurre raramente. Impresión basilar se caracteriza por invaginación de los márgenes del agujero occipital hacia arriba en el cráneo, lo que resulta en la protrusión de la apófisis odontoides dentro del foramen magno. Impresión basilar puede progresar a la compresión del tronco cerebral, hidrocefalia obstructiva o siringomielia debido a la obstrucción mecánica directa del flujo del LCR normal. Impresión basilar puede causar dolor al toser, neuralgia del trigémino, la pérdida de función de las extremidades, o parestesias. Al nadar, los individuos con impresión basilar pueden percibir que la temperatura del agua se diferencia por debajo y por encima del ombligo. Signo de Lhermitte (sensación de hormigueo en la flexión del cuello) se puede demostrar en cualquier momento. En sus niveles más severos de la participación, se pueden presentar apnea del sueño y de la muerte. Impresión basilar sintomática requiere una cirugía correctiva. El intelecto es normal, a menos que ocurra una hemorragia intracerebral.
Diagnóstico Prenatal: Por ultrasonido del feto durante el segundo trimestre. Al realizar análisis directo de la estructura de los genes o por análisis de colágeno sintetizado por células cultivadas. Al ser el diagnóstico por ultrasonido fidedigno hasta la semana 20-22 del embarazo, el diagnóstico directo se prefiere cuando se sabe que la mutación o los efectos de la misma pueden ser detectados.
Tratamiento: Los objetivos de la misma es proveer el mejor estilo de vida con una vida productiva a los individuos afectados.
-OSTEOGÉNESIS IMPERFECTA TIPO IV (MODERADAMENTE SEVERA)
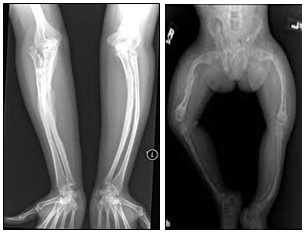
Se hereda con carácter autosómico dominante similar al Tipo I pero, con escleróticas normales. Estos pacientes nacen con fracturas e incurvaciones de los huesos largos de los miembros inferiores. Esta enfermedad parece ser heterogénea y puede asociarse a alteraciones en los dientes (tipo IV A) o no (tipo IV B). El colágeno anormal no permite la maduración de la cortical ósea de modo que, la cortical está compuesta por hueso primitivo y pequeñas áreas de hueso laminar, con los años (adolescencia o más tarde) la cortical madura. Las radiografías revelan osteoporosis, ensanchamiento metafisario y compresiones vertebrales. Son tratados con medidas ortopédicas y rehabilitación, presentan una disminución del colágeno Tipo I, muestran dentinogénesis imperfecta, escleróticas blancas y no hay sordera. Suele presentar cifoescoliosis y laxitud ligamentosa.
Tipos recientemente descritos de OI.- Los siguientes subtipos de OI siguen en la clasificación numérica Sillence. Ellos han sido subcategorized sobre la base de la histología ósea y ciertas características clínicas que ayudan diferenciación.
Este esquema de clasificación es probable que cambie con la investigación adicional sobre la fisiopatología de la OI.
-OI tipo V.- Las características clínicas distintivas de OI tipo V incluyen la ocurrencia frecuente de la luxación de la cabeza radial / subluxación y la calcificación de la membrana interósea entre el radio y el cúbito. Estos cambios no son congénitas sino que se desarrollan con el tiempo y que no se pueden encontrar en todos los Escriba pacientes V. Los individuos afectados tienen con frecuencia la formación de callo hiperplásico. Un patrón irregular de lamellation hueso se observó en el microscopio de luz polarizada. Aunque la patogénesis molecular no se ha dilucidado, es importante tener en cuenta en este momento que electroforesis colágeno es normal en estos individuos que sugiere que una proteína asociada dentro de la matriz es más probable afectada. OI tipo V representa alrededor del 5% de las personas con OI.-OI tipo VI.- OI tipo VI tiene de una moderada a grave fenotipo asociado con fracturas frecuentes, pero es clínicamente indistinguible de OI tipo IV. El sello distintivo es la mineralización ósea anormal en el examen histológico.
-OI tipo VII y VIII .- Estos subtipos de OI se distinguen por herencia autosómica recesiva. La desregulación de la hidroxilación postraduccional del residuo prolina en la posición 986 de COL1A1 ha sido implicada en la patogénesis de estos tipos de OI. Esta hidroxilación alfa depende de tres proteínas; proteína de cartílago asociada (CRTAP), prolil-3-hidroxilasa-1 (LEPRE1) y Cyclophylin B, que forman un complejo activo en el recticulum endoplásmico. Las mutaciones en CRTAP han sido asignadas por ser causante de la OI VII, mientras que las mutaciones en LEPRE1 que codifica prolil-3-hidroxilasa-1 se han asignado a OI VIII, aunque estas tareas parecen tentativas. A la fecha, no hay mutaciones patógenas en Cyclophylin B se han descrito en asociación con OI. Las características clínicas de estos dos subtipos son dependientes de las proteínas funcionales restantes y la superposición en la gravedad clínica con OI II y III OI. Hay algunos indicadores clínicos que sugieren el diagnóstico dentro del espectro de OI moderadamente grave, y puede por lo tanto ser difícil diferenciar estos subtipos. Los bebés tienden a tener fracturas congénitas y una desmineralización significativa de sus huesos. , «Palomitas de maíz,» epífisis anormales se han descrito y especulado que el resultado de la hidroxilación anormal de colágeno de tipo I en el cartílago. Este hallazgo no es patognomónico de este tipo de OI como se ha visto en las familias con aparente OI tipo III, ya que puede ser que cuando esos individuos con el tipo aparente III OI y epífisis palomitas someten CRTAP o LEPRE1 análisis de la mutación se encontrará que no tienen OI tipo III.
Las personas afectadas con OI recesiva tienden a ser de baja estatura y rizomélico con coxa vara y la esclerótica blanca. Las poblaciones identificadas inicialmente para albergar mutaciones en CRTAP o LEPRE1 eran los africanos, afro-americanos y las poblaciones de viajeros irlandeses, pero otros grupos étnicos están representados también. Aún no está claro qué porcentaje de OI es causada por CRTAP o LEPRE 1 mutaciones.
Alélica trastornos del tejido conectivo.- Ha habido varios pacientes descritos con hiperlaxitud articular y laxitud de la piel, un fenotipo clínico más acorde con el síndrome de Ehlers Danlos, pero que tienen una mayor predisposición a las fracturas. Las mutaciones no afectan a los residuos de glicina en la triple hélice de COL1 y mutaciones que implican los residuos de cisteína se han descrito en un subgrupo de estos pacientes. También se considera que las mutaciones N-terminal en consecuencia COL1 en el plegamiento de la proteína anormal, lo que conduce a un fenotipo Danlos OI / Ehlers más leve.
Consideraciones clínicas generales. Rasgos faciales.- Los bebés y los niños con OI a menudo se describen como teniendo una cara en forma triangular. El cráneo es relativamente grande en comparación con el tamaño del cuerpo y la prominencia frontal se observa con frecuencia. Platibasia se asocia comúnmente con la impresión basilar, una característica presente en aproximadamente el 11% de los individuos con los tipos III y IV de OI.
El prognatismo También se ha informado con frecuencia. El matiz de la esclerótica se hizo hincapié en las clasificaciones anteriores; esta característica es, sin embargo, la variable marcadamente y se limita a un artefacto de defectos de colágeno como un todo. Se cree que resulta de una combinación del aumento de la opacidad de los tejidos esclerales que permiten diferentes grados de pigmentación de la retina para ser visualizados y Modificación de las propiedades de difracción de luz de las proteínas de la matriz anormales. Aunque esta característica debe alertar al diagnosticador a la posibilidad de un defecto de colágeno subyacente, no siempre es útil para diferenciar los subtipos de OI.
Dientes.- DI se caracteriza por dientes translúcidos, descoloridos que son frágiles. Ellos entran en erupción antes que en años normales emparejados poblaciones de control. Si los caducifolios son normales, los dientes permanentes también será normal, lo que indica que DI es inherente a los dientes y no una forma secundaria adquirió anomalía. Se cree que la dentina anormal como resultado de la desregulación de ADAMTS2 y COL1A1.
Piel.- Fácil aparición de moretones es una característica generalizada de la fragilidad del tejido conectivo y así puede ser una característica de la OI.
Audición.- Pérdida de audición conductiva y neurosensorial mixta aflige a la mayoría de los adultos con OI. Infancia La pérdida de audición afecta aproximadamente al inicio de un 7% de los niños afectados entre las edades de 5 y 9; hipoacusia pospuberal progresiva es más típico en OI. La pérdida inicial de audición conductiva resulta de las fracturas de los huesos del oído medio con la contractura y la cicatrización del yunque. Con el envejecimiento, la pérdida auditiva neurosensorial agrava el elemento conductor preexistente. La fijación del estribo no es a diferencia de la otosclerosis y técnicas quirúrgicas tales como estapedotomía utilizado para tratar la otosclerosis han demostrado un éxito similar en el tratamiento de la pérdida de audición en OI. Es interesante observar que en un modelo animal, los cambios en OI cápsula ótica murino son similares a los observados en la otosclerosis. Estos cambios se han observado junto con cambios concomitantes en BMP3/NFKB1 expresión génica, una avenida para la exploración de futuras terapias. Algunas ganancias a corto plazo se han descrito con implantes cocleares. La terapia con bifosfonatos no se ha demostrado que influyen en la pérdida de audición.
Ojos.- La agudeza visual es generalmente normal, pero hay una predisposición a la lesión en el ojo tanto espontánea y traumática. Espontánea ruptura y uveal escleral prolapso se ha documentado y una conciencia de la posibilidad de lesión en el ojo puede ser enfatizado. Hueso escleral se utiliza a menudo como un indicador de diagnóstico para la OI. La variabilidad de este hallazgo está marcada y en riesgo de evaluación subjetiva. Se ha sugerido para estandarizar el color escleral usando el sistema de Munsell, pero el valor de este enfoque sigue sin determinarse. No es inusual que los niños no afectados para exponer esclerótica azul hasta aproximadamente 15 meses de edad.
Gastrointestinal.- Aunque las quejas de estreñimiento son comunes en niños y adultos con OI que se desplazan en sillas de ruedas, no está claro si esto es una complicación de la OI en sí o del modo de transporte. La obstrucción intestinal puede ocurrir como resultado de la protrusión acetabular pero parece ser poco común.
Cardiopulmonar.- Insuficiencia pulmonar es la causa más frecuente de fallecimiento en OI tipo II y afecta a un número de individuos con tipo más severo III OI. Tanto la patología de la pared torácica y cifoescoliosis severa asociada con la compresión vertebral pueden contribuir a la enfermedad pulmonar restrictiva. Esto puede progresar a la hipertensión pulmonar y cor pulmonale posterior, lo que requiere la ayuda del oxígeno. También se han reportado prolapso de la válvula mitral y dilatación de la aorta con o sin regurgitación. Un informe reciente indicó cifras tan altas como 95% con regurgitación valvular, pero esto rara vez causa morbilidad grave a menos que están agravando los riesgos cardiovasculares. Aunque tanto el auricular y / o rotura de la aorta se han reportado en OI; no está claro si se producen con mayor frecuencia en personas con OI que en la población general. Una minoría de individuos con OI también se ha informado con disección de la arteria coronaria o aneurismas cerebrales, ambos de los cuales son relativamente comunes en la población general, la asociación de este modo sigue siendo poco clara. La movilidad reducida y la rueda de la dependencia silla pueden predisponer a la estasis vascular y un aumento del riesgo de neumonías.
Trastornos musculoesqueléticos.- La talla baja es una característica de todos los tipos de OI, incluso en su representación más leve que la estatura adulta se redujo sólo marginalmente por las normas familiares. En las presentaciones más graves, crecimiento vertical se cae de las curvas de crecimiento para el final del primer año de vida y la velocidad de crecimiento real es más lenta que la población general. Una rara asociación con espondilolistesis secundaria a pedículos alargados se ha descrito en el contexto de la OI.
Desarrollo.- Los retrasos en el desarrollo motor grueso son comunes en asociación con hiperlaxitud articular importante y deformantes tipos de OI, pero esto se puede mejorar con la edad. Desarrollo en otras áreas suele ser normal. El aumento de la circunferencia de la cabeza es más notable entre las edades de 2 y 3. La presencia de invaginación basilar (BI) y sus secuelas neurológicas se han descrito anteriormente (véase el Tipo III OI).
Esperanza de vida.- Los niños más severamente afectados con OI tipo II normalmente no sobreviven el período neonatal. La esperanza de vida para la OI I y IV es normal. En OI III, la esperanza media de vida se puede acortar debido a cifoescoliosis severa y la forma torácica anormal con enfermedad pulmonar restrictiva operadora y la insuficiencia cardiaca.
Embarazo.- La fertilidad es normal en OI. Para la mayoría de las mujeres que tienen OI, el embarazo no presenta complicaciones. La excepción se da en aquellas mujeres con OI que son muy pequeños y requieren prematuro por cesárea debido a un compromiso respiratorio. Los niños con OI son con mayor frecuencia por cesárea como consecuencia de la presentación no del vértice. No se encontraron diferencias en la frecuencia de complicaciones entre el parto vaginal o por cesárea, laxitud articular puede aumentar, como lo hace con las mujeres no afectadas, y reducir la movilidad en las pequeñas mujeres moderadamente afectadas. El sangrado no es probablemente más común de lo habitual y complicaciones de desgarro vaginal durante el parto no son comunes. No está claro si el relajamiento pélvico postparto es más común de lo habitual. No hay resultados adversos del embarazo se han registrado hasta la fecha en la mujer con OI usando bisfosfonatos durante el embarazo.
Bioquímica sérica.- Las concentraciones séricas de vitamina D, calcio, fósforo y fosfatasa alcalina son típicamente normal, sin embargo, este último es a menudo elevado en respuesta a la fractura y de la fosfatasa alcalina rara vez puede disminuir en OI.
El establecimiento de un diagnóstico.- Al igual que con cualquier trastorno, una historia clínica detallada es un elemento crucial de la evaluación diagnóstica. Esto, en combinación con un examen físico enfocado hacia las anomalías observadas en los trastornos del tejido conectivo y un examen radiológico de las ayudas esqueleto en la formulación de un diagnóstico.
La siguiente lista de características sería típica en una presentación individuo con OI.
- La historia familiar de fracturas OI o recurrentes
- Fracturas con un mínimo o ningún trauma en ausencia de otros factores, como el abuso físico infligido u otros trastornos conocidos del hueso
- La talla baja o estatura más corta de lo previsto sobre la base de la estatura de los miembros de la familia no afectados, a menudo con deformidad ósea
- Escleróticas azules
- DI
- Pérdida de audición progresiva pospuberal
- La laxitud ligamentosa y otros signos de alteración del tejido conectivo
- Los antecedentes familiares de osteogénesis imperfecta, por lo general consistentes con herencia autosómica dominante
- Las fracturas de distintas edades y etapas de la curación, a menudo de los huesos largos, pero también pueden implicar las costillas y el cráneo. Las fracturas de chips metafisaria característicos de maltrato físico infantil pueden ser vistos en un pequeño número de niños con OI
- «Bacalao» vértebras, que son la consecuencia de las fracturas por compresión de la columna vertebral, visto principalmente en el adulto
- Huesos wormianos, definidos como «huesos suturales que son 6 mm por 4 mm (diámetro) o más grande, de más de 10 en total, con una tendencia a la disposición en un patrón de mosaico.» Huesos wormianos sugieren, pero no patognomónico, para la OI. Están presentes en hasta el 60% de los niños afectados
- Protrusión acetabular, en la que la cavidad de la articulación de la cadera es demasiado profunda y el acetábulo sobresale dentro de la cavidad de la pelvis causando protuberancia intrapélvica del acetábulo
- La baja densidad ósea detectada por absorciometría de rayos X de energía dual (DEXA). Parece haber una correlación entre la DMO y el riesgo de fractura en OI
Nota:
- La densidad ósea puede ser normal, sobre todo en OI tipo I, como DEXA mide el contenido mineral en lugar de las propiedades del colágeno. DEXA, sin embargo, no es una medición cuantitativa directa como la DEXA Z -puntuación refleja la cantidad y la disposición estructural del contenido mineral en la matriz ósea, que es cualitativamente anormal en los tipos II, III, IV y OI.
- Un importante factor determinante de la densidad ósea puede ser la capacidad del individuo afectado a deambular.
- Normas de densidad ósea de los niños no está disponible todavía, pero universalmente con un mayor uso de esta modalidad en los niños, DEXA está ganando popularidad como una modalidad útil en el diagnóstico y seguimiento de las OI.
- Un estudio selectivo del esqueleto debe solicitarse.
- AP y lateral de los huesos largos se examinan para la calidad del hueso en general, haciendo una reverencia, y las fracturas de curación.
- AP y lateral de la columna vertebral se examinan para cifoescoliosis y / o compresiones vertebrales.
- Dos o tres vistas del cráneo también se deben obtener para detectar huesos wormianos.
Diagnóstico diferencial.- El diagnóstico diferencial de la OI está determinado en gran medida por la edad de presentación y la gravedad clínica.
-En el útero
Ecografía prenatal de OI severa puede conducir a una consideración de hipofosfatasia, displasia tanatofórica, displasia campomélica y acondrogénesis ya que todos los presentes con la macrocefalia relativa y acortamiento rizomélico de las extremidades. La aparición de las manos tiende a ser normal en OI. Ecografistas experimentados suelen tener poca dificultad en la identificación de los huesos largos y costillas arrugados cuentas que diferencian OI tipo II a partir de estos otros trastornos. Ventriculomegalia se observa a menudo en OI.
La infancia y la niñez
-Trauma no accidental (abuso infantil)
OI tiene que ser distinguida de maltrato físico infantil / trauma no accidental. La prevalencia de maltrato físico es mucho mayor que la prevalencia de la OI y se ha informado que sólo el 7% de los niños con fracturas inexplicables tienen una condición médica subyacente. En raras ocasiones, ambos pueden ocurrir en el mismo niño. La historia del paciente, los antecedentes familiares, el examen físico, las imágenes radiográficas, y el curso clínico, todo ello contribuye a la distinción de OI de abuso infantil. La superposición de las características clínicas incluye múltiples o recurrentes fracturas, fracturas que no coinciden con la historia del trauma, y el hallazgo de las fracturas de distintas edades y en diferentes etapas de curación. La presencia continuada de las fracturas en un niño que ha sido retirado de una posible situación abusiva se presta apoyo a la posibilidad de OI. Metafisarias y costillas fracturas, que se consideran prácticamente patognomónico de abuso de los niños, pueden presentarse en OI. La presencia o ausencia de escleróticas azules no es fiable para distinguir OI de abuso infantil porque escleróticas azules se encuentran a menudo en los bebés normales no afectados hasta unos 15 meses de edad, también los niños con OI tipo IV y formas recesivas de la OI pueden no tener escleróticas azules.
Los antecedentes familiares son a menudo revelan nada; familias sospechosas de posible abuso infantil a menudo proporcionan un historial familiar no verificada de fracturas frecuentes, por el contrario, la historia familiar de las personas con OI a menudo no revela ninguna otra individuos afectados debido a una mutación de novo en el caso índice o la presencia de un fenotipo leve en los familiares. Las pruebas de laboratorio (estudios basados en proteínas o una prueba genética molecular de COL1A , COL1A2 y potencialmente LEPRE y CRTAP ) a menudo no es necesaria, y en algunos casos, el tiempo necesario para llevar a cabo dichas pruebas pueden retrasar la disposición adecuada de los casos de abuso infantil.
–Hipofosfatasia Infantil (OMIM 2415000) es un trastorno autosómico recesivo que se presenta en la infancia con un defecto osteogénico caracterizado por micromelia, ramas arqueadas y osteoporosis severa, incluyendo un cráneo poco mineralizada. La esclerótica son a menudo azul y las fracturas no son infrecuentes. La distinción de OI severa se ve facilitada por la observación de que la bioquímica de suero es anormal en hipofosfatasia. Hipofosfatasia infantil está causada por mutaciones en el tejido no específico de fosfatasa alcalina, y por lo tanto, los niveles de la enzima son bajos en asociación con pirofosfato de alta inorgánica, mientras que los niveles de fosfatasa alcalina tienden a ser normal o elevada en OI debido a la mayor tasa de recambio óseo.
-Síndrome de Bruck (OMIM 259450) es una enfermedad autosómica recesiva caracterizada por la fragilidad ósea, contracturas articulares congénitas, pies zambos, escleróticas normal o azul, y los huesos wormianos. Es el resultado de defectos en la lisilhidroxilasa (PLOD2) que hidroxila los residuos lisil amino-terminales implicados en la formación reticular.
Alélica trastornos del tejido
-Síndrome pseudoglioma Osteoporosis (OMIM 259770) incluye la fragilidad ósea y fracturas, otras deformidades esqueléticas, pseudoglioma con ceguera en la infancia, y otras anomalías. Es causada por mutaciones en el gen que codifica la proteína relacionada con el receptor de lipoproteínas 5.
-Síndrome de Cole-Carpenter (OMIM 112240) es caracterizado por deformidades óseas, fracturas múltiples, proptosis ocular, órbitas poco profundas, craneosinostosis orbital, prominencia frontal, y la hidrocefalia.
–Síndrome Hadju-Cheney (OMIM 102500) se caracteriza por baja estatura, retraso del crecimiento, pérdida de audición conductiva, rasgos dismórficos, la pérdida prematura de dientes, anomalías genitourinarias, osteopenia, fracturas patológicas, huesos wormianos, fracaso de la osificación de la sutura, impresión basilar, anomalías vertebrales , cifoescoliosis, la inestabilidad cervical, laxitud articular, luxación de la cabeza radial, fíbulas inclinó largo, pseudoclubbing, dígitos de corta distal, acroosteólisis, y el hirsutismo.
-Osteodisplástica gerodermia (OMIM 231070) se caracteriza por enanismo, piel laxa, la osteoporosis, los huesos wormianos, fracturas por compresión vertebral, el aspecto facial arrugado debido a la laxitud de la piel del rostro.
-Osteoporosis juvenil idiopática.- Osteoporosis juvenil idiopática se presenta típicamente en preadolescentes con fracturas y osteoporosis. La susceptibilidad a la fractura y osteoporosis por lo general se resuelven espontáneamente con la pubertad. La etiología de la osteoporosis juvenil idiopática es desconocida.
-Dentiogenesis imperfecta.- DI puede ocurrir por separado de OI como una condición familiar aislada como resultado de mutaciones en el DSPP gen en el cromosoma 4.
Diagnóstico.- Se diagnostica principalmente en los hallazgos clínicos y radiológicos. La densitometría es una ayuda de diagnóstico cuando falta la clínica y la radiología. Niños sanos que tienen más de 3 fracturas de baja energía en un año, deben ser sometidos a densitometría ósea, como parte de un rastreo de OI. Niños con OI tienen a nivel del cuello femoral disminuida la BMD (Bone Mineral Density) en comparación con niños sanos, de la misma edad y peso. Se ha postulado que los pacientes con OI tienen deficiencias en la mineralización, secundariamente a las anomalías de la síntesis del tipo I del colágeno.
El diagnóstico se puede confirmar mediante pruebas genéticas bioquímicas y / o molecular en algunos casos. Además puede ser útil hacer estudios de colágeno tisular que se realizan con una biopsia de perforación del hueso. Se aprecia una disminución del colágeno Tipo I (que forma las laminillas óseas a nivel de la piel) y mayor proporción del colágeno Tipo III, en todos los tipos de osteogénesis imperfecta (véase las Figuras 2 y 3). Los análisis bioquímicos suelen dar resultados normales aunque, muchas veces se descubre una hiperfosfatasemia sobretodo del tipo ácido y un aumento en la excreción urinaria de mucopolisacáridos e hidroxiprolona pero carece de significación para el diagnóstico diferencial, pues son síntomas secundarios de osteopatía.
Una vez conocido el diagnóstico molecular específico, a los miembros de la familia se les puede hacer una prueba por medio de un examen de sangre para ADN.
En la mayoría de las personas afectadas de OI, los rayos x se convierten en una prueba frecuente y necesaria de asistencia al diagnóstico y tratamiento. Hay peligro para la salud con la exposición frecuente a los rayos x, directamente relacionada con la intensidad de éstos. Es conveniente guardar un registro con las fechas en las que se le han realizado radiografías.
-Bioquímica sanguínea: Proteína, pruebas bioquímicas.- Históricamente, las pruebas de bioquímica fue la primera prueba de la línea para confirmar un diagnóstico clínico de la OI. El advenimiento de las tecnologías de secuenciación se ha desplazado el foco, y ahora es posible llevar a cabo el colágeno y no colágena secuenciación de genes como un enfoque de primera línea, sólo la investigación de las propiedades bioquímicas de los colágenos esta prueba debe ser negativo. El colágeno análisis bioquímico y análisis de la mutación tienen sensibilidad y especificidad similar, aunque la secuenciación tiene la ventaja de ser capaz de confirmar los defectos en todos los cuatro genes implicados en la causa de la OI. Análisis de colágeno puede ser normal o anormal en CRTAP o LEPRE pacientes de mutación.
Análisis de colágeno procede como sigue: fibroblastos derivados de una biopsia de piel se cultivan in vitro para proporcionar una fuente de proteínas para el análisis electroforético de colágeno tipo 1. Los colágenos se etiquetan y se ensayaron en SDS-PAGE para evaluar su movilidad electroforética. Proteínas anormales migrarán de forma diferente en el gel en comparación con las muestras de control.
La sensibilidad de la prueba bioquímica es del 87% en las formas no letales de OI y alrededor del 98% en forma letal. La sensibilidad varía con fenotipo clínico y refleja el pequeño efecto de algunos de COL1A1 o COL1A2 mutaciones en el tipo de estructura y cantidad de colágeno I. Una prueba de detección que pueden ser a la vez específico y sensible para la identificación de OI sin deformación es la relación de la fosfatasa alcalina para el inositol fosfato fosfato, pero esto no es de uso generalizado
Alélica trastornos del tejido-Análisis de la secuencia de Gene
El análisis de secuencia de COL1A1 y COL1A2 ADNc para detectar mutaciones en la secuencia de codificación y análisis de la secuencia de COL1A1 y COL1A2 de ADN genómico para detectar mutaciones que alteran cualquier secuencia o la estabilidad de ARNm identificar entre 80 y 85% de mutaciones en el colágeno de tipo I genes. Las mutaciones en la mayoría de las familias son únicas, y sólo unas pocas mutaciones recurrentes (principalmente dinucleótidos CpG) se ven en más de una familia. Tasa de detección de mutaciones varía según el tipo de OI relacionadas COLA1-, una amplia base de datos de todas las 850 mutaciones conocidas se ha publicado junto con una correlación genotipo-fenotipo.
Para los pacientes en quienes se identifica ninguna mutación del colágeno tipo I, la secuencia de las CRTAP y LEPRE1 está disponible comercialmente y es un siguiente paso razonable en la secuenciación secuencial para identificar una mutación. La importancia de la identificación de estas mutaciones es establecer un riesgo genético para los futuros hijos como los padres de una persona afectada estarían en un 25% de riesgo de concebir otro hijo con la enfermedad y que sería imprescindible para que puedan ser asesorados con precisión y adecuadamente de los servicios prenatales disponibles para ellos.
El ADN genómico puede ser aislado a partir de leucocitos periféricos en la sangre o de cualquier otra muestra de tejido del individuo que está siendo probado.
Mecanismos moleculares (véase. Displasias óseas, en este mismo capítulo)
Genotipo-fenotipo.- Los estudios estructurales analizar los efectos de las sustituciones de glicina con más voluminosos, cadenas laterales de aminoácidos más hidrófilos en conjunto de triple hélice de colágeno han demostrado alteración
progresiva de la triple hélice y ensamblaje molecular. Esto está en consonancia con la teoría predicho que la mayor es la interrupción de el conjunto de la triple hélice, más grave es el fenotipo. Un reciente informe pone de relieve que las mutaciones letales ocurren en ocho grupos, que están espaciadas regularmente a lo largo de las dos terceras partes de la cadena. La gravedad de las mutaciones dentro de estas agrupaciones es independiente de la sustitución real de aminoácidos, ya que dan lugar a la omisión de exón y por lo tanto dependen más en la posición específica. El «modelo regional» de mutaciones letales se ha propuesto para identificar tales mutaciones y la asignación teórica de fenotipo es correcta 86% del tiempo. Estos dominios parecen ser sitios importantes para la interacción entre proteoglicanos y la microfibrillas de colágeno.
Las mutaciones que afectan a los enlaces disulfuro, que afectan a la estabilidad del péptido C se han demostrado que el resultado en el fenotipo de tipo IV de OI más suave. Este tipo de mutación puede resultar en la formación de un homotrímero COL1A1.
Diagnóstico prenatal.- La prueba de ADN en muestras prenatales de vellosidades coriónicas puede ayudar a hacer el diagnóstico durante el embarazo. La Osteogénesis Imperfecta severa se puede detectar por medio de un ultrasonido prenatal a las 16 semanas de gestación.
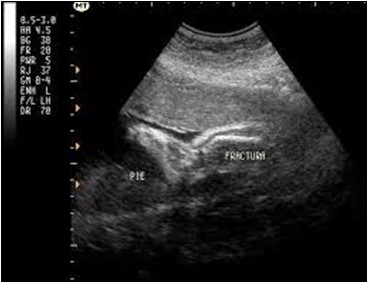
Tratamiento
1º.- Rehabilitación y terapia física.- Su principal objetivo es maximizar la función motora, especialmente importante durante la infancia.
Los resultados de la rehabilitación mejor documentados, han sido en una serie pediátrica de OI holandesa32 durante un seguimiento de 4 años de niños de 5-19 años; observaron que el rango de movilidad de la articulación disminuía significativamente a lo largo del tiempo en aquellos con OI tipo I, especialmente en los miembros inferiores, mientras que los tipos III y IV tenían limitaciones motoras más graves que no cambiaban con el tiempo. Los niños con OI tipo I no tenían manifestaciones cardiacas o pulmonares en reposo, mientras que aquellos con Tipo III o IV tenían reducida la tolerancia al ejercicio así como la fuerza muscular, lo que contribuía a la mayor fatiga durante las actividades de la vida diaria. Los niños con OI tipo I y IV que participaban en programas de entrenamiento físico de baja resistencia, tenían un aumento del consumo pico de O2, mayor fuerza muscular y capacidad después de 3 meses; pero estas mejorías disminuían 6 meses después de finalizado el programa, lo que sugiere que el ejercicio regular con la intensidad correcta es importante para mejorar sus capacidades. Para los niños inmovilizados un estudio Pilate de vibración de cuerpo entero usando una tabla inclinada en 4 niños tipo III –IV permitió sentarse a 2 niños y caminar con mínimo apoyo a otros 2, los cuales nunca habían mejorado durante varios años de tratamiento con bisfosfonatos.
2º.-Cirugía ortopédica.- Continúa siendo una piedra angular del tratamiento a largo plazo de la OI y es complementaria de la rehabilitación física. Las osteotomías de los huesos largos con colocación de clavos intramedulares, corrigen la deformidad ósea que impide una función adecuada. La cirugía correctiva es a veces fundamental para conseguir la deambulación. Actualmente los cirujanos tienen a su disposición 2 tipos de clavos telescópicos, los sistemas intramedulares telescópicos de Fassier-Dubal y los clavos intramedulares telescópicos de Sheffield que son una nueva versión de los clavos de Dubow-Baile también conocidos como los clavos que no se elongan y que permiten la inmovilización de los clavos largos, tras los procedimientos de osteotomía. Los clavos de Fassier-Dubal tienen la ventaja de su colocación percutánea, minimizando el trauma, permitiendo la reparación de varios huesos en la misma sesión y realización posterior de una rehabilitación precoz.
3º.-Tratamiento farmacológico.- Basándose en que en la OI está incrementado el turnover óseo, Devogelaer et al., tratan por primera vez a un niño con OI con pamidronato disódico (Kampar®; Aminomux®), un potente antiresortivo óseo por vía oral. Años más tarde los estudios histomorfométricos realizados por Glorieux et al. demostraron que en la OI se asocia al aumento de la actividad osteoclástica, una reducción en la formación de hueso, hecho posteriormente confirmado por otros autores. De estos hallazgos se deducirían dos opciones terapéuticas reducir la actividad osteoclástica y favorecer la formación ósea.
a) Reducir la actividad osteoclástica:
- -Bisfosfonatos
- -Inhibidores del RANKL
- -Otros futuros tratamientos: Inhibidores de Catk.
Bisfosfonatos: Desde 1987, en que se publica el primer tratamiento a un niño con OI hasta la aparición de la publicación por Glorieux et al. en 1998, que sienta la recomendación de la terapia con bisfosfonatos, este tratamiento estaba muy limitado a pequeños grupos de pacientes. A partir de esta publicación varios autores han descrito su experiencia en el tratamiento con pamidronato IV de pacientes con OI, y más recientemente el tratamiento oral con bisfosfonatos. En los últimos años se está utilizando en los niños zoledronato IV (Lezomiv®), Zometa®), bisfosfonato con ventajas sobre el pamidronato (Aredia®): al ser más potente se necesita menos cantidad y dura más el efecto, lo que permite alargar el intervalo de tiempo entre los ciclos, se administra en única dosis por ciclo y en menor tiempo.
Los bisfosfonatos son fármacos antirresortivos, ampliamente administrados a los niños con OI. Se han observado efectos positivos en la histología del hueso que incluyen un aumento del número de trabéculas y engrosamiento cortical, así como un aumento de los Z Scores vertebrales en el DXA. Los estudios en niños han demostrado que las ganancias son máximas en los 2-4 primeros años del tratamiento. Los estudios controlados demuestran que los bisfosfonatos mejoran la geometría vertebral; pero no se ha demostrado disminución de fracturas en los huesos largos, incluso en ensayos con más de 125 niños.
Estudios en ratones y en humanos han mostrado cierta preocupación por las altas dosis acumuladas en el hueso, la alteración de la remodelación ósea, la disminución de la calidad material del hueso, la mineralización y la alteración de las células óseas, sin embargo la osteonecrosis de la mandíbula no ha sido demostrada en pacientes con OI. Debido a que los bisfosfonatos tienen una vida media de más de diez años en el hueso, es crucial determinar la menor dosis acumulada efectiva para mejorar la geometría vertebral.
Por este motivo en nuestra unidad desde el 2005 lo ponemos a menor dosis y en 27 horas, siendo los resultados obtenidos similares a cuando poníamos más dosis y durante 3 días. Existe experiencia cambiando el tratamiento a zoledronato a nuestros niños mayores de 5 años, que habían recibido pamidronato previamente y se han iniciado directamente el tratamiento con zoledronato a otro de 5 años con aplastamientos vertebrales. Las dosis utilizadas han sido de 0,05 mg/Kg/dosis (máximo 4 mg) y disminuyendo la dosis a la mitad si en el ciclo anterior tuvo hipocalcemia severa, se disuelve en 50 c.c de suero salino, y se administra IV en 45 minutos, en dosis única que se repite cada 6 meses. La administración de calcio y vitamina D se inicia 1 semana antes del ciclo y la prolongación después de él, depende de los controles de calcio iónico. Tras 64 ciclos administrados, hemos observado que los que ya habían recibido tratamiento previamente con pamidronato no tuvieron ningún efecto secundario en el primer ciclo y el que no lo había tenido, tuvo un pico febril de 41º a las 24 horas del primer ciclo. A las 48 horas de administrar el ciclo se presenta una elevación de la PTH que duplica o triplica la previa al ciclo (lo cual no se ha observado con pamidronato ni cuando se lo administra en 3 días con más dosis o con el protocolo actual) y una disminución del calcio sérico en todos, el iónico solo en 2 casos.
Efectos secundarios del tratamiento con pamidronato: en general son leves y pasajeros, el más frecuente un cuadro pseudogripal en el 1er ciclo. En bebes se puede añadir un componente de broncoespasmo. La hipocalcemia asintomática es también frecuente en cualquiera de los ciclos. Otros efectos: dolor en zona de la administración por flebitis y gastroenteritis, sobre todo en el 1er ciclo.
Un inconveniente de los fármacos antiresortivos es que disminuyen la actividad de la modelación y remodelación ósea, un descenso sostenido durante el crecimiento de la remodelación ósea, puede resultar perjudicial, al poderse acumular residuos de cartílago de crecimiento en el tejido óseo trabecular, este cartílago calcificado tiene una alta densidad mineral (aumenta el valor de la densitometría) pero es menos resistente y se puede fracturar; por otra parte la baja actividad remodeladora puede retrasar la regeneración ósea tras la realización de osteotomías en los enclavamientos intramedulares, esto hace que se recomiende retrasar el ciclo de tratamiento en 4-6 meses tras estas intervenciones; pero no tras una fractura. La aparición de fracturas en las zonas de unión del hueso tratado y no tratado las hemos observado en los tratamientos interrumpidos con bisfosfonatos.
Inhibidores del RANKL: Denosumab (DMAB) (Prolia®, Xgeva®)
El Denosumab es un anticuerpo monoclonal que se une al RANKL, impidiendo la activación de su receptor RANK, presente en la superficie de los osteoclastos y otras células inmunitarias, esta unión inhibe la formación, función y supervivencia del osteoclasto, lo que lleva a una disminución de la resorción ósea en el hueso cortical y trabecular. Denosumab demostró por primera vez su efectividad para reducir la actividad osteoclástica y aumentar la masa muscular en mujeres posmenopáusicas, y en el año 2010 se autorizó su uso para el tratamiento de la osteoporosis. Los nuevos conocimientos acerca de la fisiopatología de OI-VI, obtenidos mediante la identificación del defecto genético subyacente, alentaron al grupo del Dr Semler a abordar terapéuticamente el bloqueo de RANKL en pacientes con este grave tipo de OI. Han iniciado el tratamiento en 4 niños con OI-VI que habían mostrado continuamente niveles elevados en orina de los marcadores de osteoclasia (resorción ósea) durante un anterior tratamiento con bisfosfonatos. Se administró denosumab por vía subcutánea a dosis de 1 mg/kg, cada 3 meses, tal y como se describió para adultos tratados con denosumab 36 horas después de las inyecciones, se comenzó a suministrar VO suplementos de calcio (750 mg/día) y vitamina D (500 u.i./día), que se prolongo durante 2 semanas. Ninguno tuvo síntomas de hipocalcemia. No obstante reseñan que un intervalo de 3 meses podría ser demasiado largo para pacientes con OI-VI. Un intervalo de 8 semanas parece más adecuado para garantizar una supresión constante de la resorción ósea.
Los motivos de la no respuesta a bisfosfonatos, todavía no se han investigado de forma experimental, pero podrían estar relacionados con el hecho de que los bisfosfonatos están ligados a la superficie ósea mineralizada e inducen la apoptosis de los osteoclastos en el momento de la resorción. El aumento de la cantidad de osteoides no mineralizados en pacientes con OI-VI posiblemente dificulte la capacidad de los bisfosfonatos para ligarse al hueso y, por ello, reduce su toxicidad para los osteoclastos. Denosumab al actuar de forma diferente evita la maduración y la activación de los osteoclastos, antes de que esas células se adhieran a la matriz ósea.
Ofrece potencialmente otra ventaja importante, en comparación a la terapia estándar actual que se utiliza para OI: el anticuerpo humanizado se degrada dentro de los 3-4 meses siguientes a la inyección y, por lo tanto, no permanece en el organismo, los bisfosfonatos se almacenan en los huesos durante años, un hecho que ha dado lugar a un debate todavía vigente acerca de su seguridad a largo plazo cuando se suministra a niños.
En el futuro, Denosumab podría utilizarse también como tratamiento alternativo para niños con otros subtipos de OI o en otras enfermedades osteoporóticas, de hecho, se ha publicado recientemente su utilización en un niño con displasia fibrosa con buena respuesta respecto al dolor óseo, los marcadores de recambio óseo y la mejoría de una lesión femoral.
Inhibidores de Catk (ODANACATIB) (MK-0822)
La catepsina K es una cisteinproteasa expresada de forma abundante y selectiva en los osteoclastos inhibiendo la resorción ósea, localizándose en los lisosomas, en el borde rugoso del osteoclasto maduro y en la laguna de resorción sobre la superficie ósea. La catepsina K tiene una función esencial en el remodelado óseo, y la inhibición farmacológica de esta enzima puede ser una futura opción terapéutica en procesos que cursan con un aumento de la resorción ósea. La cuantificación sérica de catepsina K abre una nueva posibilidad para conocer el nivel de actividad osteoclástica, y la monitorización de sus cambios, en pacientes que siguen un tratamiento antiosteoporótico, para valorar la efectividad del tratamiento. Este fármaco está siendo desarrollado por la farmacéutica Merck & Co., y se encuentra en Fase III.
b) Favorecer la formación ósea.
-PTH, no está indicada en niños.
-GH, incrementa el remodelado óseo, ya incrementado en estos niños.
Otros posibles fármacos: Inhibidores de la serotonina. Inhibidores de la esclerostina
Tratamientos futuros:
Terapia génica
Transplante de médula, en fase experimental.
Inhibir la expresión del gen mutado: Introduciendo ribozimas o nucleótidos antisentido que anulen el producto del gen mutado, dejando intacto el producto del gen normal (se convertirían los tipos graves en formas leves, con menos colágeno pero normal).
Los nuevos conocimientos acerca de la fisiopatología de OI, obtenidos mediante la identificación del defecto genético subyacente, permitirán en un futuro disponer de más opciones terapéuticas para estos niños.
-Evaluaciones iniciales y proyecciones de los controles
- Auditiva: deberían ser examinados por la evaluación formal de audiología periódicamente para evaluar la gravedad. Cirugía del Estribo ha demostrado ser beneficioso en varias series y los implantes cocleares han demostrado ser prometedores como los resultados son similares a los resultados obtenidos con otras causas de la pérdida auditiva neurosensorial.
- Imagen para BI: Hay una alta frecuencia de BI en pacientes con OI severa, que generalmente es lentamente progresiva en la infancia. Los pacientes que presentan temprano con síntomas menores tienen buenos resultados a largo plazo con la cirugía de descompresión ventral. Se recomienda que los niños con síntomas de ser referidos para evaluación neuroquirúrgica y la TC y la resonancia magnética. Janus et al informó que la resonancia magnética periódicas no influyeron en el proceso de toma de decisiones clínicas.
- Alélica trastornos del tejido
- Dental: evaluación dental temprana es importante y la participación de un ortodoncista es esencial para la salud dental de los individuos que presentan DI. La reconstrucción de dientes con un barniz compuesto se ha demostrado que mejora la estética y la función en los adultos jóvenes con DI. Evaluación dental debe realizarse siempre antes de los bifosfonatos se prescriben para los niños que tienen la dentición.
Pronóstico.- Se puede presentar deformidad permanente de las extremidades y puede ocurrir daño cerebral debido a las fracturas de cráneo. Dicho trastorno puede ser fatal y se clasifica según el tipo:
- Tipo I. Compatible con una expectativa normal.
- Tipo II. La mayoría de las personas afectadas, aunque no todas, mueren en etapas tempranas de la niñez.
- Tipo III. Deformidad progresiva con reducción de la expectativa de vida.
- Tipo IV. Compatible con una expectativa de vida normal.
En todos los tipos se pueden presentar también problemas cardiovasculares de diferente pronóstico. A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por Osteogénesis Imperfecta es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal, dentro de las limitaciones que imponga el grado de movilidad de cada uno.
Complicaciones.- Neumonía recurrente.
- Falla cardíaca (cor pulmonale).
- Lesión cerebral.
- Deformidad permanente.
- Problemas de respiración.
- Pérdida de la audición.
Bibliografía
- Antonella Forlino, PhD; Wayne A. Cabral, BA; Aileen M. Barnes, MS; Joan C. Marini, MD, PhD CME Released: 06/14/2011; Valid for credit through
- Osteogenesis Imperfecta Foundation: Fast Facts. Consultado el 05-07-2007.
- Dr. Martin Etchart. Capítulo 12. Anatomía Patológica Osteoarticular (artículo completo disponible en español). Pontificia Universidad Católica de Chile. Escuela de Medicina. Último acceso 25 de junio de 2008.
- Esteban C y García-Planells J. «Osteogénesis imperfecta» (en español). Genagen (Web de Interés Sanitario).
- Rauch F, Glorieux FH (2004). «Osteogenesis imperfecta». Lancet 363 (9418): pp. 1377–85. doi:10.1016/S0140-6736(04)16051 0.
- Asociación de Huesos de Cristal de España (AHUCE). Osteogénesis Imperfecta (OI) | Fundación AHUCE
- Rauch F & Glorieux FH. Osteogenesis Imperfecta. Lancet 2004; 363:1377-1385.
- Sillence, D. O. & Rimoin, D. L. Classification of osteogenesis imperfecta. Lancet 1978;1: 1041-104
- Prockop, D. J. & Kivirikko, K. I. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem 1995; 64: 403-434.
- Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci 2005;118: 1341-1353
- Willing MC, Deschenes SP, Slayton RL & Roberts EJ. Premature chain termination is a unifying mechanism for COLIAI null alleles in osteogénesis imperfecta type I cell strains. American Journal of Human Genetics 1996; 59:799-809
- Byers PH. Osteogenesis imperfecta: perspectives and opportunities. Current Opinion in Pediatrics 2000; 12:603-6097.-
- Byers PH Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest 2002; 109:3-6.
- Glorieux FH, Rauch F, Plotkin H et al. Type V osteogénesis imperfecta: a new form of brittle bone disease. J Bone Mineral Res 2000; 15: 1650-1658.
- Glorieux FH, Ward LM, Rauch F et al. Osteogénesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Mineral Res 2002; 17: 30-38.
- F. S. Van Dijk, G.Pals, R.R. Van Rijn (Classification of Osteogenesis Imperfecta revisited. Eur J Med Genet 2010;53:1-5.
- Viljoen D, Versfeld G, Beighton P. Osteogenesis imperfecta with congenital joint contractures (Bruck syndrome). Clin Genet. 1989;36:122-6.
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, Bruckner-Tuderman L, Curry CJ, Pyott S, Byers PH, Eyre DR, Baldridge D, Lee B, Merrill AE, Davis EC, Cohn DH, Akarsu N, Krakow D. Mutations in the gene encoding the RER protein FKBP65 cause autosomalrecessive osteogenesis imperfecta. Am J Hum Genet 2010;86: 551-559.
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007;39: 359-365.
- Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Jandar CY, Becker J, Breer S, Altunoglu U, Stricker S, Krawitz P, Hecht J, Schinke T, Makareeva E, Lausch E, Cankaya T, Caparrós-Martin JA, Lapunzina P, Temtamy S, Aglan M , Zabel B, Eysel P, Koerber F, Leikin S, Garcia KC, Netzer C, Schönau E, Ruiz-Perez VL, Mundlos S, Amling M, Kornak U, Marini J, Wollnik B. 2013. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet, submitted.
- Forlino, et al. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011; 7:540-557.
- Martínez-Glez V, Valencia M, Caparrós-Martín JA, Aglan M, Temtamy S, Tenorio J, Pulido V, Lindert U, Rohrbach M, Eyre D, Giunta C, Lapunzina P, Ruiz-Perez VL. (2012). Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat 2010;33(2):343-50.
- Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013 Feb 23.
- Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C. A Mutation in the 5’-UTR of IFITM5 Creates an In-Frame Start Codon and Causes Autosomal-Dominant Osteogenesis Imperfecta Type V with Hyperplastic Callus. Am J Hum Genet 2012;91(2):349-357.
- Asharani PV, Keupp K, Semler O, Wang W, Li Y, Thiele H, Yigit G, Pohl E, Becker J, Frommolt P, Sonntag C, Altmüller J, Zimmermann K, Greenspan DS, Akarsu NA, Netzer C, Schönau E, Wirth R, Hammerschmidt M, Nürnberg P, Wollnik B, Carney TJ. Am J Hum Genet. 2012;90(4):661-7.
- Morello, R., Bertin, TK, Chen, Y., Hicks, J., TonaChini, L., Monticone, M., Castagnola, P., Rauch, F., Glorieux, FH, Vranka, J. , et al. (2006). Se requiere CRTAP para prolyl 3-La hidroxilación y las mutaciones causan una osteogénesis imperfecta. Célula 127, 291-304.
- Lindert, U., et al. «Mutaciones MBTPS2 causan defectuoso intramembrane proteólisis regulada en la osteogénesis imperfecta ligada al cromosoma X,» Nature Communications, 6 de julio de 2016.