INFORMACIÓN BÁSICA.- Síndrome de Dyggve-Melchior-Clausen (DMC) (ORPHA:239).– El Síndrome de DMC es una displasia esquelética del grupo de las espondiloepimetafisarias de aparición evolutiva.
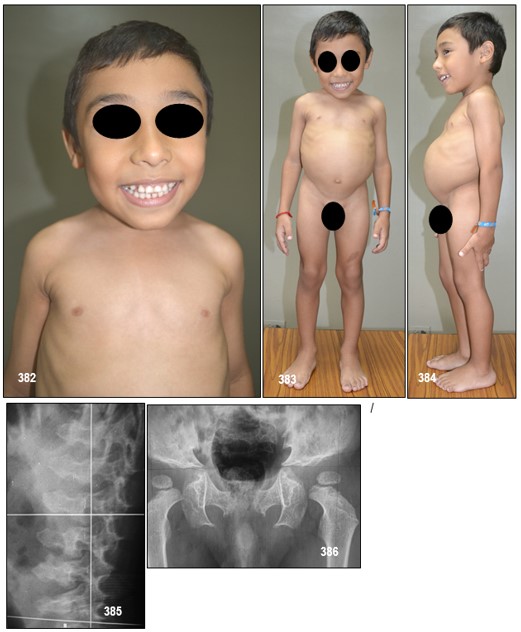
El Síndrome de DMC se caracteriza por múltiples anomalías esqueléticas que dan lugar a enanismo con microcefalia, mentón ancho y prominente, con un aspecto facial característico, platispondilia acentuada y progresiva, tronco corto con tórax ancho, redondeado y protuberante, acortamiento rizomélico de las extremidades, cifoscoliosis, alas ilíacas con irregularidades que le dan aspecto de «encaje» y retraso mental variable, que suele ser también progresivo.
Las primeras manifestaciones suelen detectarse a partir del primer año, aunque en algunos casos empezaron a apreciarse antes.
El síndrome de DMC es una entidad autosómica recesiva, cuyo gen, dymeclin (DYM), se ha localizado en el cromosoma 18q12-q21.
Este síndrome se considera muy poco frecuente, habiéndose calculado su frecuencia en 0,1:10 niños nacidos vivos.
Sin embargo, se cree que puede estar infradiagnosticado, ya que muchos casos se consideraban seudo-Morquio, o bien quedaban sin diagnóstico. Sólo hemos encontrado 2 trabajos sobre casos españoles de este síndrome en la literatura médica referenciada en las bases internacionales: uno descrito en 1976 y otro del año 2005, en el que sólo se presentan los resultados de la resonancia magnética de una niña de 8 años y 4 meses de edad, de la que no se describen las características clínicas en detalle ni su procedencia.
Aunque en un trabajo previo sobre los resultados moleculares se ha incluido una breve descripción del caso que aquí se presenta, nos parece importante publicarlo completo en nuestro país, junto con la revisión de los últimos hallazgos, por varios motivos: a) porque al ser un síndrome tan poco frecuente no es fácil su diagnóstico precoz; b) porque hoy día ya es posible su diagnóstico molecular, y c) porque al ser un síndrome de afectación multisistémica los pacientes van a ser atendidos por diversos profesionales (pediatras, neurólogos, psicólogos, internistas, ortopedas, radiólogos, fisioterapeutas y médicos de atención primaria) y, dada la dificultad diagnóstica del síndrome, cabe la posibilidad de que sean objeto de multitud de análisis y pruebas, que nada tendrían que ver con su enfermedad.
Ésta es una situación demasiado frecuente en alteraciones que, como este síndrome, son raras y que pueden evitarse si se conocen.
Epidemiología.-. Han sido reportados solamente 58 casos en todo el mundo, principalmente en Líbano (por lo que en ocasiones se describe el síndrome como “cara de libanés”), Marruecos, Groenlandia y dos casos en India.
Es una entidad poco frecuente, presentando similitudes fenotípicas con la enfermedad de Morquio y se ha intentado demostrar la existencia de alteraciones metabólicas de los mucopolisacáridos. En la literatura a nivel mundial no hay datos concluyentes y actualmente no existe un marcador bioquímico específico; hasta el momento, sólo se han descrito síntomas, signos clínicos y radiológicos de este síndrome.
Herencia- El síndrome DMC es una displasia esquelética progresiva rara del tipo espóndilo epimetafisiaria, con una forma autosómica recesiva (MIM 223800) y otra recesiva ligada al X (MIM 304950), descrita por Yunis en 1980 en una familia colombiana. La primera, que es la que ocupa este reporte, es producida por la pérdida o mutación que afecta la función del gen DYM, mapeado en 18q12- 21.
La forma que presenta herencia autosómica recesiva se produce por una mutación que causa la pérdida o disminución de la función del gen dymeclin (DYM), el cual codifica una proteína del mismo nombre, de 669 aminoácidos, de la cual no se conoce bien su función específica. Han sido reportadas al menos 21 mutaciones distintas del mismo.4 Las células con mutaciones del gen DYM muestran múltiples defectos en el transporte citoplasmático que se evidencian por agrandamiento y dispersión del retículo endoplasmático y aumento del número de las vesículas de la membrana intracitoplasmática, lo que provoca un trastorno de almacenamiento.
Genética.- El gen DYM, responsable del síndrome de DMC, se localiza en la región q12.1- q21.1 del cromosoma 18 y está compuesto por al menos 17 exones, para los que se han identificado, al menos, 21 mutaciones. Se ha estimado que tiene aproximadamente 400 kb, aunque la longitud del exón 1 aún no se ha determinado, porque el lugar de inicio de la transcripción no se ha definido.
La mayoría de mutaciones identificadas en el gen predicen una pérdida de función de su producto. DYM se expresa en la mayoría de tejidos y codifica para la dimeclina, una proteína que interactúa con las membranas del aparato de Golgi, pero de la que se desconoce su rol en la célula. La transmisión es autosómica recesiva.
Este gen ha mostrado estar muy conservado a lo largo de la evolución desde las plantas al ser humano. El producto del gen es una proteína que tiene 669 aminoácidos y que es necesaria para el desarrollo normal del esqueleto y de la función cerebral, aunque se desconocen su naturaleza y función exacta. Por los resultados de algunos estudios parece que el gen se expresa en una gran variedad de tejidos (hueso, cerebro, corazón, páncreas, próstata, pulmón y riñones), y hay evidencias de que la pérdida de función del gen podría ser el mecanismo que da lugar a la patogenia del síndrome.
En la actualidad, tras la identificación del gen, se ha confirmado que los síndromes de DMC y de Smith-McCort (SMC) están causados por mutaciones en el mismo gen DYM, por lo que se consideran alélicos. La mayoría de las mutaciones identificadas en el síndrome de DMC predicen una pérdida de la función del gen, pero las encontradas en los niños diagnosticados de SMC son mayoritariamente mutaciones de cambios de aminoácidos (missense mutation), que originan una proteína disfuncional.
Esta diferencia en la proteína del síndrome de SMC ha dado lugar a que se considere que esta mutación presumiblemente podría estar asociada a una actividad residual del gen DYM y, por ello, a un fenotipo menos grave. Sin embargo, en una publicación reciente se describe el estudio molecular de 3 familias en las que los padres eran consanguíneos; 2 de ellas habían tenido 2 hijos con el síndrome de DMC cada una, y en la tercera había sólo un hijo diagnosticado de síndrome de SMC. El análisis molecular confirmó el diagnóstico de los casos con síndrome de DMC, pero el niño con síndrome de SMC no tenía ninguna mutación del gen DYM ni presentaba la histología típica de los fibroblastos, que sí se observó en los niños con síndrome de DMC. Estos resultados llevaron a los autores a considerar que el síndrome clínico denominado SMC, aunque sea clínicamente homogéneo, debe de tener heterogeneidad genética.
Un aspecto interesante del estudio de las mutaciones en pacientes de diferentes poblaciones es la posibilidad de indagar el origen de algunas de ellas. En este sentido, Cohn et al identificaron, en pacientes procedentes de la isla de Guam, una mutación consistente en una transición A _ G que impide la transcripción del exón 8, lo que da lugar a una terminación prematura de la síntesis de la proteína. Paupe et al habían observado esta misma mutación en un paciente cuya familia era de origen español. Con posterioridad, en un trabajo publicado recientemente por Pogue et al se observó la misma mutación que afecta al exón 8 en 3 pacientes, uno procedente de Chile, otro de Argentina y el tercero de España. Mediante el estudio de SNP (single nucleotide polymorphism) se observó que había un solo haplotipo asociado con esta mutación en las familias de la isla de Guam, lo que invita a pensar que la mutación que afecta al exón 8 proviene de un antepasado común. En ese trabajo se consideró que, puesto que la expedición de Magallanes había pasado por Chile, Argentina y la isla de Guam, es probable que esta mutación y el haplotipo relacionado en las familias de la isla de Guam sean de origen español-portugués, y que fueran los integrantes de la expedición de Magallanes los que llevaran la mutación alrededor del mundo en el siglo XVI.
Esa hipótesis implicaría que el síndrome de DMC tendría que ser más frecuente en España y Portugal. Por otra parte, dado que el gen DYM, aunque con expresión variable, tiene un efecto en diversos sistemas orgánicos (sistema nervioso central, esquelético, etc.), los pacientes serán atendidos por diversos especialistas médicos, que al no encontrar un diagnóstico concreto, o llegar incluso a uno incorrecto, podrían realizar múltiples pruebas que no estarían relacionadas con el síndrome. Por ello, creemos que es importante darlo a conocer a todos los profesionales sanitarios, no sólo porque podría ser más frecuente en España de lo que cabe deducir de los casos descritos, sino, más importante aún, para ejercer lo que hoy empieza a denominarse «prevención cuaternaria», que consiste en evitar o atenuar la intervención médica innecesaria en el diagnóstico, tratamiento y/o prevención; situación ésta particularmente frecuente en los síndromes con afectación polisistémica y de frecuencias tan bajas que constituyen el paradigma de las enfermedades raras.
Frecuencia.- La forma más común de esta enfermedad es la variante ligada al cromosoma X, con una prevalencia estimada de 1 entre 150.000-200.000.
En nuestro país al carecer de estadísticas específicas, sería el primer caso publicado.
Manifestaciones clínicas.- Los pacientes se caracterizan por presentar un tronco corto y ancho, “en forma de tonel, protrusión esternal, cifoscoliosis, platiespondilia severa, huesos iliacos irregulares, acortamiento rizomélico de los miembros, microcefalia, cara con rasgos grotescos y un grado variable de retraso mental. Esta condición es extremadamente rara y su diagnóstico se hace difícil, si no existe una experiencia previa con casos similares.
Radiología.- Evidencias radiológicas que revelan una platispondilia con doble ondulación de los cuerpos vertebrales, displasia epifisaria y metaepifisaria y crestas ilíacas festoneadas.
Diagnóstico.- Esta condición es extremadamente rara y su diagnóstico se hace difícil, si no existe una experiencia previa con casos similares.
El síndrome de DMC puede sospecharse después de una evaluación clínica completa, un historial detallado del paciente y la identificación de los hallazgos característicos (p. ej., tórax de barril y desproporción) baja estatura). Las radiografías pueden confirmar anomalías esqueléticas específicas y hallazgos compatibles con el síndrome de DMC. Prueba genética también puede confirmar un diagnóstico.
Estudios con microscopia electrónica muestran la presencia de vacuolas membranosas citoplásmicas muy grandes y anormales, así como un retículo endoplásmico granular alargado y abundante.
Diagnóstico diferencial.- El síndrome Dyggve-Melchior-Clausen tiene similitudes clínicas con otras entidades lo que provoca confusiones diagnósticas iniciales y necesariamente se debe realizar diagnóstico diferencial en primer lugar con la forma ligada al cromosoma X (MIM 304950), con la mucopolisacaridosis tipo IV o síndrome Morquio (MIM 253010), desorden lisosomal debido a la deficiencia de las enzimas N-acetilgalactosamina6-sulfatasa o la beta-galactosidasa, 6 una variante más rara como la displasia de Smith-McCort (MIM 607326), la displasia espóndilo epifisiaria tarda (MIM 313400), y la displasia espóndilo epifisiaria congénita (MIM 183900).
Estos síndromes suelen poseer una clínica similar pero con signos radiológicos y enzimáticos específicos. Algunos autores plantean la posibilidad de el DMC se trate de una mucopolisacaridosis y aportan datos bioquímicos, sin embargo hasta el momento no se tienen resultados concluyentes. Actualmente la presencia de los signos clínicos y radiológicos y ausencia de mucopolisacariduria se consideran como criterio diagnóstico de DMC.
Tratamiento.- El tratamiento de individuos con DMC depende de los síntomas y generalmente es de apoyo. No hay cura para esta condición. Los tratamientos pueden incluir la fusión espinal de los segmentos de la columna vertebral en la parte superior de la columna vertebral u otros medios de estabilización vertebral. Se pueden usar técnicas quirúrgicas adicionales para corregir diversas anomalías esqueléticas, como la dislocación de las articulaciones del hombro y la cadera. En algunos casos, se requiere reemplazo de cadera.
En este sentido, la sustitución protésica total, precedida o no de osteotomías correctoras, será necesaria en la mayoría de los pacientes a edades más tempranas que en la población general.
Posibles actuaciones quirúrgicas:
- Osteotomía correctora de coxa vara: Ángulo de inclinación del fémur mide unos 130 grados fisiológicamente; si este ángulo se cierra hablamos de coxa vara; si se abre hablamos de coxa valga. En cualquiera de los dos casos hay que hacer osteotomía (cortes) para abrirla o cerrarla respectivamente. Hacemos una varizante (en caso de coxa valga) o valguizante (en caso de coxa vara) según el caso.
- Endoprótesis en artrosis de cadera
- Fusión vertebral en hipoplasia odontoides.
Pronóstico.- Los niños con síndrome de DMC pueden beneficiarse de la intervención temprana y programas educativos especiales.
El síndrome de DMC es una condición progresiva, lo que significa que los signos y síntomas empeoran con el tiempo. Las complicaciones ortopédicas generalmente progresan, lo que puede incluir lordosis lumbar, cifosis torácica, escoliosis, dislocación en las articulaciones de la cadera, rodillas deformadas y compresión de la médula espinal. La altura adulta está severamente reducida con una altura que varía de 82 cm a 128 cm (32 a 50 pulgadas). Las complicaciones neurológicas pueden incluir hiperactividad, comportamiento de tipo autista, falta de habla de leve a grave. Discapacidad intelectual.
Bibliografía
- Dyggve HV, Melchior JC, Clausen J. Morquio-Ulrich’s disease: an inborn error of metabolism? Arch Dis Child. 1962;37:525-34.
- Yunis, E. Fontalvo, J., Quintero, L. X-linked Dyggve-Melchior-Clausen syndrome. Clin Genet. 1980;18:284-290. 1.
- Osipovich AB2. , Jennings JL, Lin Q, Link AJ, Ruley HE. Dyggve-Melchior-Clausen syndrome: chondrodysplasia resulting from defects in intracellular vesicle traffic. Proc Natl Acad Sci U S A. 2008 Oct 21;105 (42):16171-6.
- Dimitrov A3. , Paupe V, Gueudry C, Sibarita JB, Raposo G, Vielemeyer O, Gilbert. The gene responsible for Dyggve-Melchior-Clausen syndrome encodes a novel peripheral membrane protein dynamically associated with the Golgi apparatus. Hum Mol Genet. 2009 May 1;18(9):1714-6.
- Paupe V4. , Gilbert T, Le Merrer M, Munnich A, Cormier-Daire V, El Ghouzzi V. Recent advances in Dyggve-Melchior-Clausen syndrome. Mol Genet Metab. 2004 Sep-Oct;83(1-2):51-9.
- Rastogi SC5. , Clausen J, Melchior JC, Dyggve HV, Jensen GE. Lysosomal (leucocyte) proteinase and sulfatasa levels in Dyggve-Melchior-Clausen (DMC) syndrome. Acta Neurol Scand. 1977 Nov;56(5):389-96.
- Galasso C6. , Fabbri F, Pagnotta G, Palusci A, Sanna ML, Serrao Arnone D, Scirè G. Dyggve-Melchior-Clausen syndrome: description of 2 further cases. Pediatr Med Chir. 1995 Nov-Dec;17(6):573-6.
- Lantigua Cruz Araceli. Introducción a la Genética Médica. Ciudad de la Habana: Editorial Ciencias Médicas, 2004. Cap.18. 7.
- Dyggve H8. V, Melchior JC, Clausen J, Rastogi SC. The Dyggve-Melchior-Clausen (DMC) syndrome. A 15 year follow-up and a survey of the present clínicasl and chemical findings. Neuropädiatrie. 1977 Nov;8(4): 429-42.
- Coëslier A9. ; Boute-Bénéjean O; Moerman A; Fron D; Manouvrier-Hanu S. Dyggve-Melchior-Clausen syndrome: differential diagnosis of mucopolysaccharidosis type IV or Morquio disease. Arch Pediatr. 2001 Aug;8(8):838-42.
- Ehtesham N10. ; Cantor RM; King LM; Reinker K; Evidence that Smith-McCort dysplasia and Dyggve-Melchior-Clausen dysplasia are allelic disorders that result from mutations in a gene on chromosome 18q12. Am J Hum Genet. 2002 Oct;71(4):947-51.
- Neumann LM11. ; El Ghouzzi V; Paupe V; Weber HP; Fastnacht E; Leenen A; Lyding S; Klusmann A; Mayatepek E; Pelz J; Cormier-Daire V. Dyggve-Melchior-Clausen syndrome and Smith-McCort dysplasia: clínicasl and molecular findings in three families supporting genetic heterogeneity in Smith-McCort dysplasia. Am J Med Genet. 2006 Mar 1; 140(5):421-6.
- Reinaldo Menéndez García,I Miladys Orraca Castillo,II Jesús Juan Rodríguez,III ldefonso Friol Sánchez,IV Madelín Rodríguez Ríos,V Lourdes Reyes Puentes.VI . Síndrome Dyggve-Melchior-Clausen: hallazgos clínicos y radiológicos en tres casos de dos familias de Pinar del Río. Rev Cubana Genet Comunit. 2012;6(3):48-53