Comentarios de los Editores: La displasia espondiloepifisiaria congénita (donde se incluye la Displasia de Kniest) pareciera ser siempre causada por una anomalía de gen autosómico dominante. Esto significa que un adulto con este trastorno tendrá una posibilidad de 50% de pasar este gen pobremente funcional a cada hijo. No es poco común que una persona con este trastorno nazca de padres de estatura promedio. Cuando esto sucede normalmente es debido a un nuevo cambio en la probabilidad (mutación) solo en el óvulo o espermatozoide que genera a la persona afectada. Aun cuando los casos de mosaicismo germinal se han reconocido en pocas ocasiones, en general esto significa que los padres de estatura promedio que han tenido un hijo con este trastorno no tienen un riesgo significativo de que vuelva a ocurrir en los siguientes hijos.
INFORMACIÓN BÁSICA.- Displasia de Kniest (OMIM:156550/ORPHAN:485/CIE-10:Q77.7).- La Displasia de Kniest es una Displasia Espondiloepifisiaria Congénita es una de un grupo de trastornos óseos y cartilaginosos que se conocen como Colagenopatías de Tipo II. Es decir, cada miembro de este grupo de trastornos (que también incluyen, por ejemplo, el síndrome de Stickler, a la displasia de Kniest, displasia espondilometafisiaria, ciertas formas de acondrogénesis, etc.) surgen debido a los cambios en la síntesis del colágeno tipo II, codificado por el gen COL2A1. El colágeno tipo II es particularmente importante en los tejidos conectivos de las articulaciones (cartílago), en el crecimiento óseo, en las vías aéreas y el desarrollo del ojo.
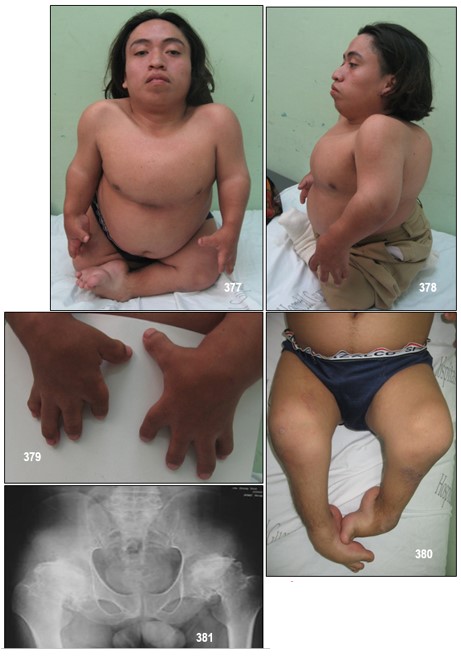
Por lo anteriormente enunciado, la Displasia de Kniest es una colagenopatía de tipo II grave, caracteriza por tronco y extremidades cortas, articulaciones prominentes e hipoplasia mediofacial (cara redondeada con raíz nasal plana). La prevalencia es desconocida. La enfermedad es evidente desde el nacimiento. Paladar hendido (a veces asociado al síndrome de Pierre-Robin, ver este término), cifoescoliosis, osteoartritis prematura, miopía grave, desprendimiento de la retina y sordera son rasgos comunes.
Los afectos no pueden apretar los puños, aparentemente debido a los reducidos espacios de las articulaciones. Otras características de la enfermedad son una elevada excreción de queratán sulfato en orina y macrocefalia.
La estatura de los pacientes es variable, pero suele ser muy inferior a la normal. La inteligencia es generalmente normal.
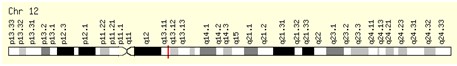
Genética.- La enfermedad se transmite de forma autosómica dominante y está causada por mutaciones en el gen COL2A1 (12q13.11-q13.2), que codifica para el colágeno tipo II. Por otro lado, el gen SOX9 (17q24) codifica para el factor de transcripción que hace que los condrocitos produzcan el colágeno tipo II normal del hueso, a través de la activación del gen COL2A1. Si alguna de las copias del gen SOX9 está mutada no se activará la transcripción del gen COL2A1 de manera que se producirá un colágeno anormal.
El COL2A1 gen proporciona instrucciones para la fabricación de un componente de colágeno de tipo II, llamado la cadena pro-alfa1 (II). El colágeno tipo II añade estructura y resistencia a los tejidos conectivos que el apoyo del cuerpo los músculos, articulaciones, órganos y piel. El colágeno de tipo II se encuentra principalmente en el cartílago, un tejido resistente pero flexible que conforma gran parte del esqueleto durante el desarrollo temprano. Más tarde cartílago se convierte en hueso, excepto por el cartílago que sigue para cubrir y proteger los extremos de los huesos y está presente en la nariz y oídos externos. El colágeno de tipo II es también parte del gel transparente que llena el globo ocular (el vítreo), el oído interno, y la parte central de los discos entre las vértebras de la columna vertebral (núcleo pulposo).
Para construir el colágeno tipo II, tres (II) las cadenas de pro-alfa1 retuercen entre sí para formar una molécula de procolágeno tipo cuerda de triple cadena. Moléculas de procolágeno se procesan entonces por las enzimas en la célula. Una vez procesadas, las moléculas abandonan la célula y se organizan en largas y finas fibrillas que enlazan entre sí (reticulación) en los espacios alrededor de las células. Los enlaces cruzados resultan en la formación de tipo II, maduros fibras de colágeno muy fuertes.
Las mutaciones en el gen COL2A1 causa una forma de acondrogénesis conocida como Tipo 2 Algunas mutaciones eliminan parte del gen COL2A1 o conducen a pro-alfa1 (II) las cadenas que faltan segmentos críticos.
Otras mutaciones cambian uno de los bloques de construcción de proteínas (aminoácidos) que se utilizan para hacer que el pro-alfa1 (II) de la cadena. Específicamente, el aminoácido glicina se sustituye con un aminoácido diferente en varios lugares de esta cadena de colágeno. Todas estas mutaciones impiden la producción normal de colágeno de tipo II maduro, lo que resulta en las anomalías esqueléticas graves se observan en este trastorno.
Al menos 18 mutaciones en el gen COL2A1 se han encontrado para causar hipocondrogénesis. Algunas mutaciones eliminar parte del COL2A1 gen o conducen a pro-alfa1 (II) las cadenas que faltan segmentos críticos. Otras mutaciones cambian uno de los aminoácidos que se utilizan para hacer que el pro-alfa1 (II) de la cadena. Específicamente, el aminoácido glicina se sustituye con un aminoácido diferente en varios lugares de esta cadena de colágeno.
Todas estas mutaciones interfieren con la formación de moléculas de colágeno de tipo II de cadena triple maduros, que dan lugar a las características de hipocondrogénesis al afectar a los tejidos que son ricos en colágeno de tipo II.
La mayoría de las mutaciones responsables de la Displasia de Kniest eliminan uno o más bloques de construcción de ADN (nucleótidos) en el gen COL2A1. Estas mutaciones causan anormalmente cortos pro-alfa1 (II) las cadenas de colágeno que se producen en la célula, que luego se unen con largas cadenas de colágeno, de longitud normal. El desajuste de pro-alfa1 (II) de colágeno cadenas resultados normales y anormalmente cortos de tipo II moléculas anormales de colágeno que son más cortos de lo habitual. Esta anormal de colágeno tipo II evita que los huesos y otros tejidos conectivos se desarrollen correctamente, lo que causa baja estatura, problemas en las articulaciones, y otros signos y síntomas de la Displasia de Kniest.
Histopatología.- El síndrome de Kniest se asocia a anormalidades en la organización de las fibrillas de colágeno, se aprecian más finas de lo normal y de aspecto irregular, sin su característico patrón bandeado. Además, se ha demostrado la ausencia de condrocalcina de la matriz extracelular de cartílagos epifisarios, y se concentran anormalmente en las vacuolas intracelulares donde no formaban parte de la molécula del procolágeno. El tamaño de la cadena alfa del colágeno de tipo II es normal, lo que indica la formación de la triple hélice, el contenido de colágeno de tipo II también es normal. Estas observaciones indican que el defecto en la displasia de Kniest se debe a la secreción de procolágeno de tipo II carente del propéptido C y a una anormal formación de la fibrilla, el propéptido C normalmente se requiere para la formación de la fibrilla.
Diagnóstico.- Se hace en base a los datos radiológicos, que incluyen: epífisis grandes y deformes, cabeza femoral ausente, metáfisis femorales superiores ensanchadas, platispondilia y otras malformaciones vertebrales.
Los estudios patológicos del cartílago muestran la presencia de inclusiones intracitoplasmáticas en los condrocitos y una matriz rica en vacuolas. Las formas de displasia espondiloepifisaria y displasia metatrópica (ver estos términos) constituyen los principales diagnósticos diferenciales.
En los pacientes jóvenes, los rasgos clínicos y radiológicos son similares a los de la displasia oto-espondilo-megaepifisaria (ver este término), pero en este último caso, la miopía está ausente. La mayoría de los casos se deben a mutaciones heterocigotas de novo del gen COL1A2, pero los padres afectados deben estar informados de un riesgo de recurrencia del 50%. El diagnóstico molecular prenatal es posible en las familias en las que la mutación responsable ya ha sido identificada. La micromelia se puede detectar durante el segundo trimestre del embarazo y el escáner TC del feto puede ser útil para el diagnóstico.
Tratamiento.- Es sólo sintomático.
Pronóstico.- Depende de la presencia de malformaciones en las articulaciones y anomalías vertebrales.
Bibliografía
- Kniest W. Zur Abgrenzung der Dysostosis enchondralis von der Chondrodystrophie. Eur J Pediatr 1952; 70: 633-40. 2. Kniest W, Leiber B. Kniest-Syndrom. Mschr. Kinderheük 1977; 125: 970-3. 3. Horton WA, Rimoin DL. Kniest Dysplasia. A Histochemical Study of Growth Plate. Pediat Res 1970; 13 : 1266-70.
- Ascurra M, Herreros M, Rodríguez S. Displasias óseas: a propósito de cuatro nosologías diferentes. Instituto de Investigaciones en Ciencias de la Salud 2005; 5: 98-100.
- Baltaxe E, Suárez F, Garante I. Displasia campomélica. Descripción de un caso. Colombia Médica 2005; 36: 266-270
- http://www.ivami.com/
- https://www.researchgate.net/publication/344445979_DISPLASIA_ESPONDILOEPIFISIARIA_O_DISPLASIA_DE_KNIEST