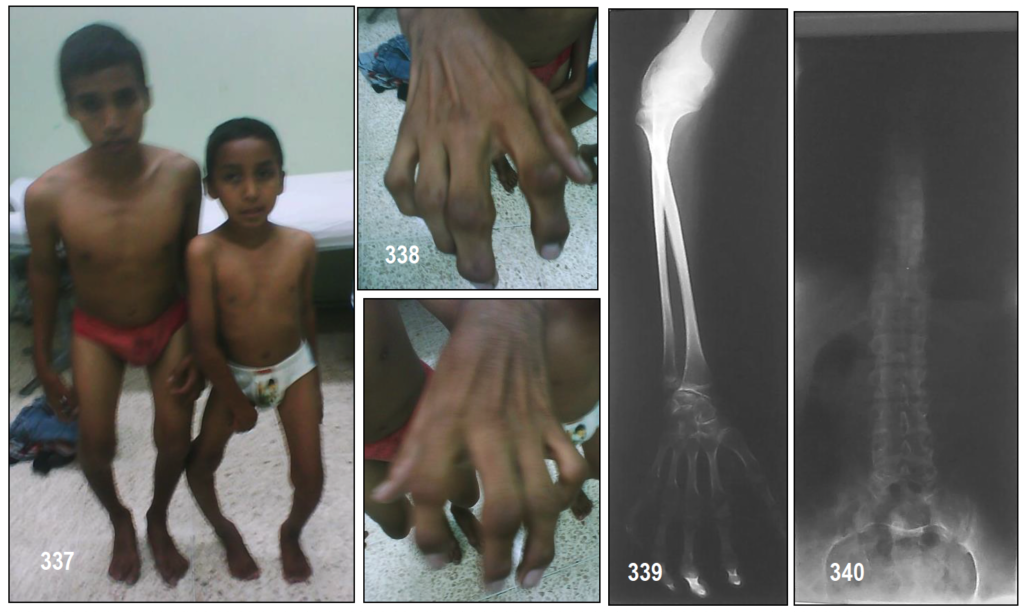
INFORMACIÓN BÁSICA.- Pseudoacondroplasia.- Camptodactilia de Tel Hashomer (OMIM 211960/ORPHA:750).- El síndrome de camptodactilia de Tel Hashomer es un síndrome raro caracterizado por camptodactilia, hipoplasia muscular y debilidad, anomalías esqueléticas, dismorfismo facial, prolapso de la válvula mitral y deformidades músculo-esquelético y dermatoglifos anómalos. En 2005 se habían descrito 20 casos. Los rasgos dismórficos incluyen: asimetría facial, hipertelorismo, puente nasal ancho, filtrum largo y boca pequeña. También se observa con frecuencia: escápula alada, escoliosis, sindactilia y clinodactilia. Los pacientes afectados suelen tener un desarrollo intelectual normal. Todavía no se conoce la base molecular del síndrome. La transmisión es probablemente autosómica recesiva. Los dos casos que presento tuvieron características consistentes con el diagnóstico de THC. El modo de herencia más probable es autosómica recesiva. Muchos de los casos antes mencionados sobre la aparición en los hermanos de consanguinidad de los padres como fue el caso con que presento. Gollop et al reportaron camptodactilia del quinto dedo de un padre que podría ser una manifestación de heterocigotos. Esta es la única familia reportado hasta el momento en que un padre tenía la expresión de la rara enfermedad.
Patton et al llevar a cabo la electromiografía (EMG) y biopsia muscular, por primera vez, que mostraron miopatía y la histología muscular mostraron tamaño anormal de fibra. La electromiografía hecho en el presente caso mostró características miopáticos. La biopsia muscular representa un fracaso embriológico en el desarrollo muscular en lugar de la enfermedad muscular progresiva. Pagnan et al habían reportado once pacientes. Ninguno de ellos tenía anormalidades del corazón. Los dos de mis pacientes presentaron prolapso de la válvula mitral y la regurgitación mitral tal como informaron Toriello et al de dos hermanos con THC que tiene prolapso de la válvula mitral.
Las características adicionales que se describen en la literatura hasta la fecha se resumen en la Tabla 24.

Clasificación desde el otro lado, camptodactilia y miopatía, deja menos diferenciales. Miopatía nemalínica se puede considerar, pero el fenotipo clínico es muy diferente.
Hay algunas características que sugieren un trastorno del tejido conectivo, así como prolapso de la válvula mitral y paladar ojival.
Las otras características tales como debilidad sintomática, los hallazgos de la electromiografía y punto hipoplasia muscular hacia el proceso miopático. Camptodactilia revela un defecto de proliferación de fibroblastos.
Síndrome camptodactilia Tel Hashomer anterior ha sido reportado en Israel, India, Rusia, Italia, Arabia y Brasil, y ahora en Ecuador, que sugiere que la reserva genética está presente en todas las poblaciones.
El Síndrome camptodactilia Tel Hashomer es una enfermedad rara que es posiblemente infradiagnosticada. Este caso pone de relieve la necesidad de que el reconocimiento de esta rara entidad de modo que más de informes de casos esporádicos y familiares afectados ayudaría en la identificación de las bases genéticas de este síndrome. Los estudios familiares ayudarían en el mapeo de genes. Técnicas moleculares modernas, como la secuenciación de próxima generación podrían allanar el camino para la delimitación de la patogenia molecular de esta enfermedad y que confirma el modo exacto de herencia.
Sería también probablemente nos dirá si la combinación de camptodactilia, miopatía y cardiopatía congénita (prolapso de la válvula mitral) es un trastorno genéticamente heterogéneo o si es alélica a uno de los trastornos de un único gen conocido. La comprensión de la fisiopatología ayudará en la gestión, incluyendo la factibilidad, los resultados y la rehabilitación de los pacientes que se someten a cirugía para camptodactilia planificación. La asesoría genética será más significativa una vez que se identificó el gen. El diagnóstico de los THC debe ser tomado en consideración cuando un paciente presenta con camptodactilia con debilidad sintomática. Como prolapso de la válvula mitral es una condición de silencio, se requiere una ecocardiografía de detección en tales pacientes. Esto ayudará a diagnosticar este síndrome muy raro y profundizar en la fisiopatología y el análisis molecular de la enfermedad.
Bibliografía
- Bueno M, Bueno-Lozano M, Bueno AL. Osteocondrodisplasias. En: Pombo M (ed). Tratado de Endocrinología Pediátrica, 2ª ed. Madrid: Díaz de Santos, 1997; págs. 331-348.
- Goodman RM, Katznelson BM, Hertz M, Katznelson MB. Camptodactilia, con hipoplasia muscular, la displasia esquelética y pliegues palmares anormales: síndrome camptodactilia Tel Hashomer. J Med Genet 1976; 13: 136-41.
- Gollop TR, Colletto GM. El síndrome camptodactilia Tel Hashomer en una familia brasileña consanguíneos. Am J Med Genet 1984; 17: 399-406.
- Patton MA, MacDermot KD, Lago BD, síndrome camptodactilia Baraister M. Tel Hashomer: informe de un caso con características miopáticos. J Med Genet 1986; 23: 268-71.
- Pagnan NA, Gollop TR, Lederman H. El síndrome camptodactilia Tel Hashomer: aportación de un nuevo caso y revisión de la literatura. Am J Med Genet 1988; 29: 411-18.
- Toriello HV, Higgins JV, Malvitz T, Waterman DF. Dos hermanos con Tel Hashomer camptodactilia y prolapso de la válvula mitral. Am J Med Genet 1990; 36: 398-403.
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H et al. Síndromes de aneurismas causados por mutaciones en el receptor de TGF-β. N Engl J Med 2006; 355: 788-98.
- TunçbilekE, Alanay Y. congénita aracnodactilia contractural (síndrome de Beals). Orphanet J Rare Dis 2006; 1: 20.
- Patel ZM, Adhia la AR. Síndrome de Tel-Hashomer camptodactilia con hirsutismo en una familia india. J Assoc Phys. la India 2004; 52: 837-8.