Displasias óseas.–Los términos displasias óseas, displasias esqueléticas y osteocondrodisplasias se refieren a un grupo heterogéneo de trastornos del esqueleto óseo que poseen la característica común de presentar una alteración generalizada del tejido óseo y cartilaginoso. Son una de las causas más frecuentes de retraso severo del crecimiento.
Es necesario distinguir una serie de conceptos, que tienden a usarse indistintamente:
- Displasia: Procesos que implican defectos generalizados del esqueleto provocados por alteraciones intrínsecas.
- Disóstosis: Procesos limitados a un segmento óseo o hueso concreto.
- Distrofia: Defectos causados por un proceso extrínseco.
Existen más de 200 displasias óseas, algunas de ellas muy poco prevalentes; La Tabl1 resume estas patologías. La frecuencia general de estas enfermedades es difícil de establecer, pero si se incluyen las formas larvadas que no llegan a diagnosticarse, su prevalencia alcanzaría un caso por cada 1.000 habitantes, siendo la acondroplasia es la más frecuente de todas ellas.
La patogénesis de estos trastornos puede atribuirse a un déficit de componente tisular normal, a un exceso de elementos mutantes, a la acumulación de metabolitos intermediarios o a irregularidades en los mecanismos que regulan el desarrollo del tejido osteocartilaginoso1. La etiopatogenia de estas enfermedades suele estar relacionada con mutaciones en los genes que codifican proteínas colágenas. Se han encontrado mutaciones en el gen COL2A1 en las condrodisplasias con afectación ocular como la displasia congénita espondiloepifisaria o el síndrome de Kniest, mutaciones del gen COL10A1 en la displasia metafisaria de Schmid y del COL9A2 en la displasia epifisaria múltiple (DEM). Otra proteína afectada frecuentemente en estas entidades es el receptor 3 del factor de crecimiento fibroblástico (FGFR3), que aparece mutado en la acondroplasia, la hipocondroplasia y la displasia tanatrópica. La mayoría de las displasias óseas siguen un patrón de herencia autosómica dominante. Sin embargo, muchas de ellas surgen por mutaciones de novo, siendo la edad paterna avanzada un factor de riesgo para ello.
La clasificación clásica es la de Rubin2, que las distingue en hipo o hiperplasias en función de la zona del hueso afecta (Figura 1). No obstante, hoy en día se prefiere agruparlas según el origen molecular de las mismas y los desórdenes genéticos hallados, un ejemplo es la clasificación de Nelson:
- •Afectación de las proteínas de la matriz del cartílago: Displasias espondiloepifisarias, DEM.
- •Afectación de los receptores transmembrana: Grupo de la acondroplasia.
- •Alteración del transporte de iones: Displasia diastrófica, acondrogénesis.
- •Afectación de los factores de transcripción: Displasia campomélica, síndrome uña-rótula.
- •Defectos en la reabsorción ósea: Osteopetrosis.
- •Defectos desconocidos: Distrofia torácica asfixiante o síndrome de Jeune, enfermedad de Caffey o hiperostosis cortical infantil.
En la práctica clínica diaria es más útil clasificarlas por zona esquelética afecta:
- a) Esqueleto axial (displasia espondiloepifisaria [DEE] y displasia metatrópica o síndrome de Kniest),
- b) epífisis (DEM, displasia epifisaria hemimélica) y
- c) metáfisis (acondroplasia, condrodisplasia tanatofórica).
A continuación y en diferentes capítulos iremos describiendo en detalles las características más importantes de las displasias óseas más prevalentes y significativas en nuestro medio.
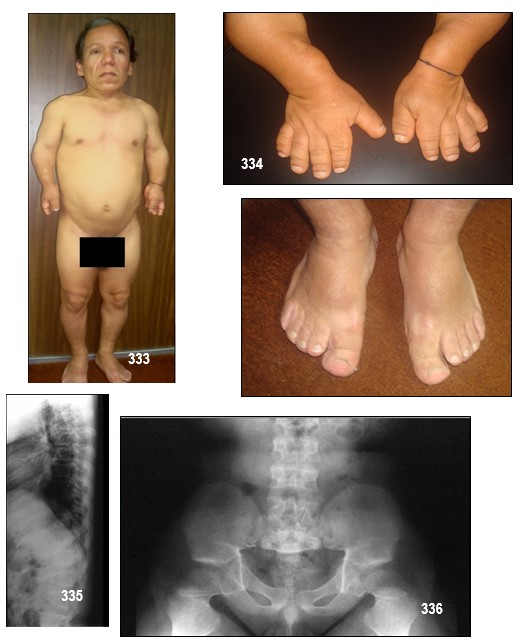
INFORMACIÓN BÁSICA.- DISPLASIAS ÓSEAS: Acondroplasia (OMIM 100800/ ORPHA:15).- La Acondroplasia (ACH) es uno de un grupo de trastornos que se denominan condrodistrofias u osteocondrodisplasias (Tabla 1). La ACH es un trastorno del crecimiento de los huesos que ocasiona el tipo más común de enanismo rizomélico congénito, se relaciona en el 75 % de los casos con mutaciones genéticas (asociadas a la edad parental avanzada) y en el 25% restante con desordenes autosómicos dominantes.
El 80% de los acondroplásicos tienen padres de estatura normal. Las parejas de adultos acondroplásicos que engendran hijos tienen un riesgo del 50% de transmitir su enfermedad, la acondroplasia heterozigótica, del 25 % de tener descendencia con acondroplasia homocigótica, que suele ser mortal por insuficiencia respiratoria, y del restante 25% de descendencia normal. La inteligencia del acondroplásico es normal. El paciente presenta mutaciones en el codón 380 del receptor del Factor de Crecimiento 3 de los Fibroblastos (FGFR- 3), lo que a su vez genera anormalidades en la formación de cartílago, y el trastorno se comporta como un carácter autosómico dominante, ya que la mayoría de los casos surge por una mutación nueva con unos padres sanos.

La ACH es una de las displasias esqueléticas más comunes con una incidencia de uno de cada 16.000 a 26.000 nacimientos vivos. Las características clínicas de ACH incluyen estatura corta rizomélico, aparente macrocefalia con hipoplasia del tercio medio facial, haciendo una reverencia de los miembros inferiores, y el aumento de la lordosis lumbar.
Las personas con acondroplasia tienen baja estatura, para los hombres la estatura adulta promedio es de 131 centímetros (51,5 pulgadas) y para las mujeres 123 centímetros (48,4 pulgadas) sin embargo la estatura puede ser tan corta como 62,8 cm (24,7 pulgadas). Una característica distintiva de este síndrome es el gibbus toracolumbar en la infancia.
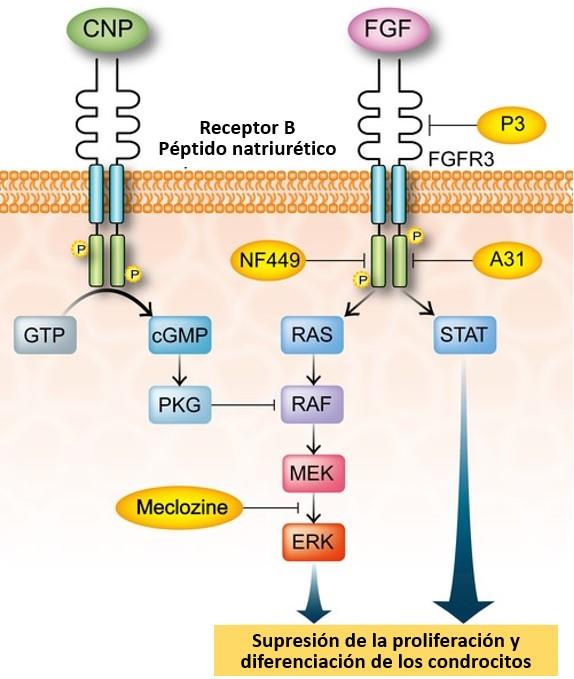
La ACH está causada por mutaciones que afectan la función del gen del receptor del factor de crecimiento de fibroblastos (FGFR-3). FGFR-3 es un regulador clave del crecimiento de hueso endocondral, que señala a través de varias vías intracelulares (Figura 22), incluyendo el transductor de señal y activador de la transcripción (STAT) y activada por mitógenos proteína quinasa (MAPK). Ganancia de función de las mutaciones de FGFR-3 causan varias displasias esqueléticas con extremidades cortas como hipocondroplasia (HCH), ACH grave con retraso en el desarrollo y acantosis nigricans (SADDAN), y la displasia tanatofórica (TD) tipos I y II. En contraste, la pérdida de función de las mutaciones en FGFR-3 alusivas al síndrome CATSHL en los seres humanos, que se caracteriza por el crecimiento excesivo del esqueleto incluyendo camptodactilia, estatura alta, escoliosis, y pérdida de la audición, así como el síndrome de cordero araña en ovejas. Estos hallazgos indican que las funciones del FGFR-3 de señalización como un regulador negativo del crecimiento de hueso endocondral.
No hay tratamientos eficaces para las displasias esqueléticas relacionadas con FGFR-3-están disponibles actualmente. La hormona del crecimiento (GH) ha sido administrado a niños con ACH basados en evidencia de un efecto beneficioso a corto plazo. La respuesta a la GH, sin embargo, es moderada y el efecto a largo plazo sigue siendo controvertido. Es concebible que la regulación negativa de la señalización FGFR-3 alivia el fenotipo esquelético de displasias esqueléticas relacionadas FGFR-3. Compuestos químicos pequeños que antagonizan la señalización FGFR3 se han identificado recientemente. Perfiles toxicológicos de estos compuestos, sin embargo, permanecen en su mayoría sin resolver. El péptido natriurético tipo C (CNP) es un potente antagonista de la señalización FGFR-3 que alivia el fenotipo corto extremidades de los ratones ACH a través de su inhibición de la vía MAPK FGFR-3. CNP tiene una vida media corta y se requiere la infusión intravenosa continua durante los experimentos in vivo. El análogo de CNP con una vida media prolongada, BMN, ha sido recientemente desarrollado y recuperación significativa del crecimiento óseo se demostró en ratones ACH por la administración subcutánea de BMN.
Se ha denunciado que algunos compuestos aprobados por la FDA para identificar un medicamento aplicable clínicamente que mejore la ACH y otras displasias esqueléticas relacionadas con FGFR-3.
Se encontró que el diclorhidrato de meclozina, un fármaco antiemético comúnmente utilizado para su actividad antihistamina, suprime eficazmente la señalización de FGFR-3 en tres diferentes líneas celulares condrocíticas y cultivo de órganos de hueso embrionario. También se identificó que meclozina suprime la fosforilación FGF-2 mediada por ERK.
Epidemiología.- Debido a que la acondroplasia es una enfermedad que tiene manifestaciones similares a otras 19 enfermedades congénitas (osteogénesis imperfecta, displasia epifisaria múltiple tardía, acondrogénesis, osteopetrosis, displasia tanatofórica, etc.), las estimaciones de su prevalencia son difíciles puesto que los criterios diagnósticos son subjetivos y cambian con el tiempo. Un ejemplo de lo anterior es que estudio detallado y de larga duración en los Países Bajos mostró que la prevalencia determinada al nacer de sólo 1,3 por cada 100.000 nacidos vivos, mientras que otro estudio realizado al mismo tiempo encontró una tasa de 1 por cada 10.000.
En otras especies.- Debido a un enanismo desproporcionado en algunas razas de perros, estos han sido clasificados como acondroplásicos. Como tal es el caso de la raza: dachshund, bassed hound y bulldog, esto solo por mencionar algunas de ellas. Datos de la Asociación de genómica, en estudios en perros con extremidades cortas, han mostrado su estrecha relación con una codificación retro genética del factor de crecimiento 4 (FGF-4).
Etiología genética.- La Acondroplasia fue mapeado en el cromosoma 4p16.3 en 1994, mutaciones y heterocigotos del FGFR3 se identificaron poco tiempo después. Mutaciones en el FGFR3 fueron descubiertos por los TDs y hipocondroplasia (véase la Figura 1). Notables grados de homogeneidad genética y genotipo: correlación fenotipo, se puso de manifiesto que prácticamente todos los pacientes con acondroplasia clásicos tenían la misma mutación Gly380Arg en el dominio de transmembrana tirosina quinasa este receptor.1, el 8 de la misma manera, todos los lactantes con TDII tenido la misma mutación Lys650Glu distal en el dominio kinasa, mientras que Asn540Lys una mutación en el dominio quinasa proximal se detectó en la mayoría de los pacientes con hipocondroplasia. Casi todos los recién nacidos con mutaciones TDI tienen que introducir los residuos de cisteína libre en el dominio extracelular de la unión ligando proximal del receptor. Cabe destacar que la mutación de la lisina 650 puede producir 3 diferentes fenotipos clínicos: la conversión a ácido glutámico resulta en TDII, la conversión de metionina a causas SADDAN, y la conversión de serina lleva a hipocondroplasia.
Patogénesis Molecular
a) Receptores (Figura 1).- El FGFR3 codifica uno de los 4 estrechamente relacionados con los receptores de FGF (FGFR1-4) en mamíferos. Todos tienen un dominio extracelular de la unión ligando, un dominio transmembrana, y un dominio intracelular que contiene una división subdominio de tirosina quinasa. Los receptores difieren en su distribución temporal y espacial de expresión.
La diversidad adicional es generada por un splicing alternativo que influye en la especificidad ligando. Las mutaciones similares a las de FGFR3 se han observado en FGFR1 y FGFR2 en humanos con síndrome de craneosinostosis.
Después de la especulación inicial de que las mutaciones de acondroplasia causan la pérdida de la función del receptor, pronto se hizo evidente que en realidad el resultado fue la ganancia en función del FGFR3 y el alcance de este beneficio se encontró en correlación con la gravedad del cuadro clínico fenotípico. Las demás pruebas vinieron de los experimentos de ingeniería genética en ratones en los cuales el FGFR3 fue inactivado o el receptor activado en el cartílago de la introducción de acondroplasia o mutaciones TD, o mediante la sobreexpresión de ligandos que activan FGFR3. Ratones en los cuales el FGFR3 fue inactivado tuvieron los huesos largos, mientras que en los ratones con un exceso de activación del FGFR3, tuvieron cortos huesos. En consecuencia, las mutaciones asociadas al FGFR3 con acondroplasia a menudo se refieren a la activación de mutaciones.
De interés es el hecho de que las funciones que se obtienen dependen de la activación de mutaciones diferentes, dependiendo del tipo de células en las que el receptor FGFR3 se expresa. Por ejemplo, la activación del FGFR3 promueve la mitosis y la diferenciación de bloques en muchos tipos de células no condrocíticas De hecho, la activación de mutaciones TD se han encontrado en el colon y carcinoma de vejiga y mieloma múltiple. En la placa de crecimiento de condrocitos, sin embargo, la activación de FGFR3 tiene el efecto contrario como se explica a continuación.
b) Dimerización.- La unión de ligandos FGF a monómeros FGFR3 conduce a la dimerización de receptores. ¿Cuál de los 22 FGFs conocidos es (son) el ligando fisiológico (s) para FGFR3 es (son) no se conoce, aunque FGFs 2, 4, 9 y 18 son probablemente los mejores candidatos sobre la base de la distribución de expresión y la capacidad para activar FGFR3 in vitro? También es concebible que diferentes ligandos FGF activen FGFR3 en diferentes situaciones fisiológicas. Sulfato de heparina- proteoglicanos en la superficie celular, como syndecans, así como splicing alternativo de subdominios unión ligando, influencian la unión específica.
La dimerización activa la intrínseca actividad de la tirosina quinasa del receptor y promueve transfosforilación de los principales residuos de tirosina en el dominio citoplásmico. Estos residuos sirven como sitios de atraque para el adaptador de señal y proteínas efectoras que son reclutados para los receptores activados y que se propagan señales FGFR3.
c) Vías de Señalización.- Señales del FGFR3 influyen en una variedad de eventos celulares y procesos en gran parte a través de inducir o reprimir la expresión de genes en una celda de contexto específico.
Cuatro principales vías de señalización han sido identificados hasta la fecha para propagar las señales del FGFR3: STAT, MAPK, PLC-g, y la PI3K-AKT (transductor de señales y activador de transcripción 1, mitogen-activated proteína quinasa, fosfolipasa C gamma, fosfato fosfatidilinositol-3 – quiinasa-serina/treonina quinasa [proteína quinasa B]) con los 2 primeros que reciben la mayoría de atención.
La mayoría de las vías de señalización se ilustra en la Figura 22. Señales del STAT1 son para inducir la expresión de inhibidores mitóticos, como inhibidor de la CDK. Uso de microarrays para evaluar los cambios en la expresión génica en las células condrocíticas, Dailey et al demostraron que señales iniciadas FGFs en múltiples vías que resultan de la inducción de las funciones antiproliferativas y por la regulación del crecimiento de la promoción de las moléculas.
Dos vías de MAPK han estado implicados, las pruebas más sólidas provienen de ratones transgénicos en los cuales la expresión constitutiva de miembros activos de las 2 vías se destinó a los cartílagos, incluyendo la placa de crecimiento del cartílago.
Expresión de MKK6 activado, que específicamente activa la vía MAPK, inhibe la proliferación de condrocitos, en parte, a través de la inducción del factor de transcripción Sox 9, la hipertrofia de condrocitos también se inhibió en estos ratones enanos. La expresión constitutivamente activa MEK1, que específicamente activa la MAPK-vía ERK, elaboró un fenotipo similar enano, a través de la inhibición de la diferenciación del condrocito terminal sin efecto inhibitorio sobre la célula en proliferación. Estas observaciones ponen de relieve la importancia de la proliferación de ambos condrocitos y terminales (hipertrófica) la diferenciación en el crecimiento lineal del hueso y el papel central del FGFR3 en la regulación negativa de estos eventos.
Es importante destacar que el FGFR3 es uno de los muchos organismos reguladores fisiológicos que modulan el crecimiento óseo lineal. Su función normal es como un regulador negativo. Las mutaciones asociadas con la acondroplasia y condiciones están relacionadas con el pensamiento de actuar a través de la exageración o la mejora de esta función fisiológica normal en lugar de la adquisición de nuevas funciones.
d) Consecuencias de las mutaciones. La causa de este trastorno es una mutación en el gen que codifica para el receptor 3 del factor de crecimiento de fibroblastos 3(FGF3), localizado en el cromosoma 4. Existen dos mutaciones posibles que afectan a este gen: G1138A y G1138C. Ambas son puntuales, donde dos pares de bases complementarias del ADN se intercambian:
–Mutación G1138A: en el nucleótido número 1138, la guanina es sustituida por adenina. En el 98% de los casos de acondroplasia, se sufre esta mutación.
–Mutación G1138C: tiene lugar el cambio de guanina por citosina, también en el nucleótido 1138. La frecuencia de esta alteración es mucho menor, apenas en el 2% de los casos.
En ambas situaciones, la repercusión en la cadena aminoacídica de la proteína FGFR-3 es la misma: el cambio del aminoácido glicina por una arginina.
Dicha mutación puede darse de dos formas distintas: por herencia autosómica dominante, cuando hay antecedentes familiares de enfermedad (alrededor del 10% de los casos) y por una mutación de novo, con padres sanos (es la causa más frecuente, hasta en el 90% de los pacientes).
Otros fenotipos asociados a mutaciones del FGFR-3 incluyen: la hipocondrodisplasia, la craneosinostosis, la displasia tanatofórica y la displasia de SADDAN. Esta última es un desorden extremadamente raro (sólo se han descrito hasta la fecha 3 casos) que se caracteriza por una estatura extremadamente corta, con tibias muy arqueadas y acantosis nigricans. Estos pacientes padecen una apnea temporal que requiere ventilación asistida.
Herencia genética.- La herencia de este trastorno es autosómica dominante, lo que significa que, para padecerlo, basta con que se herede el gen mutado de, al menos, uno de los progenitores. Las posibilidades genotípicas y sus correspondencias fenotípicas, son las siguientes:
- -Homozigoto (G1138A/G1138A): es la forma más grave del trastorno y suele ser letal durante el período neonatal. Para que tenga lugar, es necesario que ambos progenitores tengan acondroplasia (heterocigotos, pues los homocigotos no sobreviven). Ocurre en el 25% de los hijos cuando ambos progenitores son acondroplásicos.
- -Heterocigoto (G1138A/alelo normal): a este genotipo se puede llegar desde tres supuestos posibles.
Si ambos padres tienen acondroplasia, la posibilidad de que el hijo sea heterocigoto para el trastorno es de un 50%. Si únicamente uno de los padres es acondroplásico, también hay un 50% de posibilidades de heredarlo. En la mayor parte de los casos heterocigóticos, el trastorno se debe a una mutación nueva, por lo que los dos padres son de estatura normal.
Mutación de novo.- En torno al 80% de los afectados de acondroplasia no tienen antecedentes familiares del trastorno. El motivo son mutaciones espontáneas o de novo (G1138A o G1138C) que afectan a la línea germinal paterna. Son, por tanto, mutaciones que ocurren en los gametos del padre durante la espermatogénesis. Estas alteraciones se dan, como su nombre indica, de forma espontánea, lo que implica un desconocimiento de su causa; sin embargo, numerosos estudios parecen constatar una relación de la mutación de novo con la edad del padre en el momento de la fecundación, de tal manera que tener más de 35-40 años parece suponer un factor de riesgo para tener un hijo acondroplásico. La frecuencia de aparición de la acondroplasia se distribuye de igual manera entre individuos de ambos sexos y de cualquier raza.
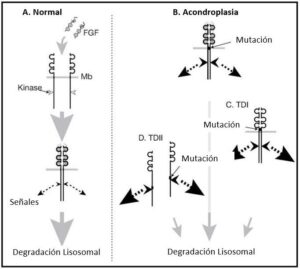
- (A) Normalmente, el ligando induce la dimerización de receptores de monómeros, que activa la cinasa e inicia la propagación de señales del FGFR-3. El FGFR-3 activado es dirigido y degradado por lisosomas relativamente poco después de la activación.
- (B) Dímeros del FGFR-3 se estabilizan por mutación (flecha) en el dominio transmembrana del receptor en la acondroplasia.
- (C) Dímeros del FGFR-3 son inducidas por la formación del disulfuro en el dominio extracelular proximal (flecha) en TDI.
- (D) La quinasa es constitutivamente activada por mutación en TDII (y, en menor medida, en SADDAN e hipocondroplasia).
- (E) La degradación Lisosomal se hace más lenta en las 3 condiciones. MB: membrana.
Causas.- Normalmente el factor FGFR-3 tiene efecto regulador en el crecimiento de los huesos. En la acondroplasia el receptor de este factor se encuentra mutado, por lo que este se encuentra constitutivamente activo lo cual lleva al acortamiento de los huesos. Las personas con acondroplasia tienen una copia normal del gen del factor FGFR-3, pero también tienen una copia mutada. Dos copias del gen mutado es fatal desde antes del nacimiento. En cuanto a la herencia genética, una persona con acondroplasia tiene el 50% de probabilidades de heredar esta enfermedad a sus hijos, lo cual significa que hay un 50% de probabilidades de que cada niño herede esta enfermedad .Por otro lado si ambos padres tienen acondroplasia, sus hijos tienen un 25% de probabilidades de morir poco tiempo después de su nacimiento, y un 50% de probabilidades de que tenga acondroplasia y un 25% de que el niño presente el fenotipo. No todas las personas que nacen con acondroplasia tienen padres con esta misma condición, ya que esto puede ser resultado de una nueva mutación. Se cree que esta enfermedad no se adquiere necesariamente por herencia genética, ya que existen nuevas mutaciones de los genes que pueden llevar a una acondroplasia, y esto está asociado principalmente con una edad avanzada de los padres (generalmente los mayores a 35 años).
Estudios actuales han demostrado que las nuevas mutaciones de los genes para acondroplasia son heredados exclusivamente del padre y que ocurre durante las espermatogénesis; (sobra) pues durante la ovogénesis existe algún tipo de mecanismo regulador que impide la mutación de los genes, sin embargo las mujeres siguen siendo capaces de presentar el fenotipo y genotipo, y (por lo tanto) de transmitir el alelo mutante. Más del 99% de (la) acondroplasia es causada por dos mutaciones diferentes del factor FGFR3.
En aproximadamente el 98% de los casos, un punto mutado G a uno A dentro del nucleótido 1138 del gen factor FGFR3 causa una substitución de la glicina por la arginina (Bellus et al. 1995, Shiang et al. 1994, Rousseau et al. 1996). El otro 1% de los casos (restantes) son causados por un punto mutado G a uno C dentro del nucleótido 1138.
El gen mutante fue descubierto por John Wasmuth y sus colegas en 1994. Existen dos síndromes que tienen una base genética similar a la acondroplasia: la hipocondroplasia y la displasia tanatofórica.
Signos y síntomas clínicos.- Las personas con acondroplasia muestran una presencia física característica como consecuencia de la interrupción del desarrollo del cartílago en las epífisis de los huesos, haciéndose más notable en los huesos largos húmero y fémur, que son los que presentan un crecimiento más rápido. De esta forma presentan una baja estatura, que no suele sobrepasar los 144 cm en la edad adulta, con acortamiento de las extremidades y agrandamiento del cráneo, mientras que el tronco conserva su tamaño normal. A continuación se muestran los hallazgos anatomoclínicos más importantes de esta enfermedad:
- -Extremidades superiores: aunque son más cortas que en un individuo normal, los antebrazos son más largos que los brazos y son incapaces de realizar una extensión completa del codo.
- – Extremidades inferiores: al igual que ocurre en los brazos, los miembros inferiores también son más cortos, con mayor evidencia en el fémur que en la tibia. El eje de las rodillas toma una postura anómala y origina el genu varo, una mayor separación entre ambas rótulas que origina unas extremidades inferiores arqueadas.
- – Signo del tridente: hace referencia a la separación existente entre el tercer (corazón o medio) y cuarto (anular) dedo de la mano.
- – Alteraciones en la columna vertebral: Durante la infancia pueden presentar una cifosis dorsal por la falta de tono muscular. En ocasiones, se combina con una hiperlordosis lumbar que intenta compensar la desviación.
La apariencia típica del enanismo acondroplásico se puede observar en el momento del nacimiento. Los síntomas pueden abarcar:
- -Apariencia anormal de las manos con un espacio persistente entre el dedo del corazón y el anular.
- -Pies en arco
- -Disminución del tono muscular
- -Diferencia muy marcada en el tamaño de la cabeza con relación al cuerpo
- -Frente prominente (prominencia frontal)
- -Brazos y piernas cortos (especialmente la parte superior del brazo y el muslo)
- -Estatura baja (significativamente por debajo de la estatura promedio para una persona de la misma edad y sexo)
- -Estenosis raquídea
- -Curvaturas de la columna vertebral llamadas cifosis y lordosis
Diagnóstico.- La acondroplasia puede ser detectada antes del nacimiento mediante un ultrasonido, el diagnóstico consiste en una ecografía fetal por discordancia progresiva entre la longitud del fémur y el diámetro biparietal por edad. Una prueba de ADN puede ser realizada antes del nacimiento para detectar la homocigosidad de la mutación, una condición que como ya se mencionó resulta letal
Pruebas prenatales.- Embarazo de alto riesgo: el embarazo de alto riesgo es aquel en el que ambos progenitores tienen acondroplasia. Las pruebas son las mismas que se llevan a cabo para el diagnóstico, utilizando ADN del feto extraído de las vellosidades coriónicas a las 10-12 semanas de gestación o por amniocentesis a las 16-18 semanas
Embarazo de bajo-riesgo: los exámenes ecográficos rutinarios pueden identificar miembros anormalmente cortos en el feto, aumentando la posibilidad de una acondroplasia. Sin embargo, estos hallazgos solo son aparentes en el tercer trimestre del embarazo. También puede analizarse el DNA fetal extraído por amniocentesis.
Se recomienda la cesárea para extraer los fetos con acondroplasia para reducir el riesgo de lesiones del SNC al pasar la cabeza por el conducto vaginal.
Hallazgos radiológicos.- Un estudio radiológico del esqueleto es útil para confirmar el diagnóstico de acondroplasia. En este estudio se puede observar un cráneo grande, con un estrecho agujero occipital y una base relativamente pequeña; cuerpos vertebrales cortos y aplanados con un espacio intervertebral relativamente grande; alas iliacas pequeñas y cuadradas con una muesca ciática estrecha y un techo acetabular horizontal; huesos tubulares cortos y gruesos con ventosas metafisarias y placas de crecimiento irregulares; crecimiento excesivo del peroné; manos anchas con metacarpos y falanges cortas y costillas cortas con forma de copa en los extremos anteriores.
- Durante el embarazo, una ecografía prenatal puede mostrar líquido amniótico excesivo rodeando al feto.
- El examen del bebé después de nacer muestra un aumento en el tamaño de la cabeza de adelante hacia atrás. Asimismo, puede haber signos de hidrocefalia.
- Las radiografías de los huesos largos pueden revelar la presencia de acondroplasia en el recién nacido.
- Si las características radiológicas no son clásicas, la búsqueda de un diagnóstico diferencial debe ser considerada.
Diagnóstico diferencial.- Aunque se conocen más de 100 displasias esqueléticas que pueden ocasionar una estatura corta, la mayoría son extremadamente raras y tienen características clínicas y radiológicas que las distinguen fácilmente de la acondroplasia.
A diferencias de otras displasias esqueléticas, la acondroplasia se encuentra presente en el momento del parto pero no está asociada a insuficiencia respiratoria.
Otras condiciones que pueden ser confundidas con la acondroplasia son la hipocondroplasia severa (también ocasionada por una mutación en el gen para FGFR3) y la condrodisplasia metafísiaria tipo McKusick
Tratamiento.- A pesar de que se conoce la mutación del gen en el receptor del factor de crecimiento, actualmente no existe un tratamiento conocido para tratar la acondroplasia. La hormona de crecimiento es usada por personas que no tienen acondroplasia para ayudarlas con su crecimiento, sin embargo esta no es efectiva en las personas que si la padecen. A pesar de ello si lo desean pueden someterse a una cirugía de alargamiento de miembros, que a pesar de ser un tema controvertido ha sido efectiva en algunos casos. Generalmente los mejores resultados aparecen dentro del primer y segundo año de la terapia con hormona del crecimiento.
Después del segundo año de terapia el efecto benéfico del crecimiento del hueso comienza a disminuir. Es por esto que la terapia con hormonas del crecimiento no tiene efectos satisfactorios a largo plazo. La terapia génica se encuentra aún en desarrollo. Una compañía de EUA, BioMarin Pharmaceutical Inc. anunció recientemente el inicio de un estudio de fase I en voluntarios sanos para BMN-111, un análogo del péptido natriurético del tipo C, para el tratamiento de la acondroplasia. Las últimas investigaciones son reguladas y controladas por la organización no lucrativa Growing Strongrer.
Recientemente el grupo de Masaki Matsushita et al., han descubierto que un fármaco aprobado por la FDA que suprime señalización de FGFR3 en ACH. Estos científicos han sugerido que la meclozina, un fármaco antihistamínico que se ha utilizado durante mucho tiempo para el mareo por movimiento, facilita la proliferación de condrocitos y mitiga la pérdida de matriz extracelular en células de condrosarcoma de rata (RCS) tratadas con FGF2.Por este y otros mecanismos evidencias en cultivos celulares, se ha propuesto que la meclozina es un agente terapéutico potencial para el tratamiento de ACH y otras displasias esqueléticas relacionadas con FGFR3.
La meclozina como un nuevo inhibidor de la señalización de FGFR3, que puede aplicarse potencialmente a la práctica clínica para la baja estatura en displasias esqueléticas relacionadas con FGFR3. La meclozina es un bloqueador H1 de venta libre que se ha utilizado de forma segura para el mareo por movimiento durante más de 50 años. Debido a que ya se han establecido las dosis óptimas y los efectos adversos de la meclozina, la meclozina se puede recetar fácilmente para la displasia esquelética relacionada con FGFR3 después de que se confirme la eficacia en humanos.
Otras terapias relacionadas con la inhibicion del FGFR3.- Jonquoyet et al. identificó que un compuesto sintético A31 es un inhibidor de la tirosina quinasa FGFR3 mediante análisis in silico . Demostraron que A31 suprime la fosforilación constitutiva de FGFR3 y restaura el tamaño de los fémures embrionarios de Fgfr3 Y367C/+ratones en cultivo de órganos. Además, A31 potencia la diferenciación de condrocitos en la placa de crecimiento Fgfr3 Y367C/+ . Jin et al. examinaron una biblioteca de fagos aleatorios de 12 péptidos y encontraron que P3 tiene una afinidad de unión alta y específica por el dominio extracelular de FGFR3. Demostraron que P3 promueve la proliferación y diferenciación condrogénica de células ATDC5 cultivadas, alivia el retraso del crecimiento óseo en rudimentos óseos de ratones TD (ratones Fgfr3 Neo-K644E/+ ) y finalmente revierte la letalidad neonatal de ratones TD. Sin embargo, estos nuevos inhibidores de tirosina quinasa FGFR3 pueden inhibir tirosina quinasas distintas de FGFR3 y pueden ejercer efectos tóxicos inesperados en humanos. La meclozina también puede inhibir las vías de la tirosina quinasa imprevistas, pero podemos predecir que no habrá ningún efecto adverso manifiesto, porque la meclozina se ha utilizado de forma segura durante más de 50 años.
CNP es otro agente terapéutico para trastornos relacionados con FGFR3. Los ratones con deficiencia de CNP quedaron empequeñecidos por el estrechamiento de las zonas proliferativas e hipertróficas de las placas de crecimiento. Las mutaciones de pérdida de función en NPR2 que codifican un receptor para CNP son responsables de la displasia acromesomélica de tipo Maroteaux (AMDM), una forma de displasias esqueléticas humanas de miembros cortos. Por el contrario, la sobreexpresión de CNP impidió el acortamiento de los huesos acondroplásicos al inhibir la vía de señalización de MAPK. Yasoda et al. demostró que la administración continua de CNP a través de infusión intravenosa normalizó con éxito el enanismo de ratones Fgfr3 ach . Como el CNP tiene una vida media muy corta, Lorget et al. desarrolló un análogo de CNP de vida media extendida en plasma, BMN111, que es resistente a la digestión con endopeptidasa neutra. Demostraron que la administración subcutánea de BMN111 exhibe una recuperación significativa del crecimiento óseo en ratones Fgfr3 Y367C/+ . La meclozina mostró una actividad inhibidora similar en la señalización de FGFR3 en comparación con CNP en cultivo de explantes óseos ex vivo , así como en células condrogénicas in vitro . Esperamos que la meclozina se pueda usar como sustituto o además del CNP y el análogo del CNP.
La vía MAPK es una de las principales vías de señalización de FGFR3 en la proliferación y diferenciación de condrocitos. La activación sostenida de ERK en los condrocitos conduce a una disminución de la proliferación, una mayor degradación de la matriz, una alteración de la forma celular y una disminución de la diferenciación. CNP inhibe la fosforilación de la quinasa RAF1 a través de la inhibición por PKGII. Masaki Matsushita et al. demostraron que la meclozina atenúa la fosforilación de ERK en los condrocitos. Gohil et al. informó que la meclozina tiene una actividad de fosforilación antioxidante (OXPHOS) además de propiedades antihistamínicas y antimuscarínicas. En su informe, la meclozina mostró actividades citoprotectoras contra la lesión isquémica en el cerebro y el corazón. Dado que otros fármacos con propiedades antihistamínicas, antimuscarínicas y anti-OXPHOS no mostraron inhibición de la señalización de FGFR3 en nuestros estudios, es poco probable que las acciones farmacológicas de la meclozina sobre la condrogénesis sean relevantes para sus propiedades antihistamínicas, antimuscarínicas y antimuscarínicas. o propiedades anti-OXFOS. Aunque se requieren estudios adicionales para demostrar que la meclozina es efectivamente efectiva para pacientes con displasias esqueléticas relacionadas con FGFR3, proponemos que la meclozina es un agente terapéutico atractivo y potencial.
Pronóstico.- Las personas con acondroplasia rara vez alcanzan 1,52 m de estatura, pero su inteligencia está en el rango normal. Los bebés que reciben el gen anormal de sus dos padres generalmente no sobreviven más allá de unos pocos meses.
Por lo tanto parece poco probable que los perros y humanos desarrollen acondroplasia por las mismas razones. Sin embargo, estudios histológicos en algunos perros acondroplásicos han mostrado alteraciones en los patrones de células en los cartílagos, lo cual es muy similar a lo observado en los humanos que presentan la enfermedad. Un tipo similar de acondroplasia se encontró en una camada de lechones daneses que tenía un fenotipo normal aparente. El enanismo fue dominante en la descendencia de la madre, y aunque los lechones nacieron fenotípicamente normales, con el tiempo se fue haciendo cada vez más evidente el padecimiento.
Conclusiones.- La tirosina-quinasa transmembrana mediada por los receptores FGFR-3 es un importante regulador negativo del crecimiento óseo lineal actuando principalmente a través de las vías de señalización STAT1, p38-MAPK, y ERK-MAPK para inhibir la proliferación y diferenciación terminal de los condrocitos en la placa de crecimiento. Las mutaciones que aumentan estas acciones producen la acondroplasia de fenotipo clínico cualitativo; el alcance de esta mejora se correlaciona con la gravedad de este fenotipo. El acto a través de mutaciones o la promoción de la estabilización de la dimerización de receptores necesarios para la activación, por activar directamente la actividad quinasa a través de cambios conformacionales del receptor y de ralentización de la degradación del receptor. Varias estrategias se han propuesto terapéuticamente para contrarrestar el aumento de la señal del FGFR-3 de salida, incluyendo químicos inhibidores de tirosina-quinasa y el bloqueo de anticuerpos, tanto selectiva para FGFR-3 como la activación de la vía CNP-NPR-B-GMPc, las cuales antagonizan las señales MAPK-ERK/p38 de abajo del FGFR-3. Las 3 estrategias han demostrado éxito en células y sistemas de cultivo de órganos, pero aún no en su totalidad los ensayos con animales, tal vez porque tiene que ser dirigidas directamente a la placa de crecimiento de los condrocitos para lograr el efecto terapéutico cada vez más restringido a los huesos.
La investigación sobre la acondroplasia y mutaciones del FGFR-3 ha estimulado mucho interés en la biología celular y molecular en lo normal y anormal del crecimiento óseo lineal. De hecho, muchos nuevos genes cuyos productos influyen en el crecimiento óseo se han descubierto en los últimos años, al igual que las vías que contribuyan a la regulación del crecimiento óseo.
La esperanza es que estos descubrimientos conduzcan a nuevos, seguros y eficaces terapias para los trastornos del crecimiento óseo lineal dentro de los próximos años.
Bibliografía
- Bueno M, Bueno-Lozano M, Bueno AL. Osteocondrodisplasias. En: Pombo M (ed). Tratado de Endocrinología Pediátrica, 2ª ed. Madrid: Díaz de Santos, 1997; págs. 331-348.
- Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, et al. (2008) The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am J Med Genet A 146A: 2385–2389. doi: 10.1002/ajmg.a.32485
- Horton WA, Hall JG, Hecht JT (2007) Achondroplasia. Lancet 370: 162–172. doi: 10.1016/s0140-6736(07)61090-3
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, et al. (1994) Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371: 252–254. doi: 10.1038/371252a0
- Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, et al. (1994) Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78: 335–342. doi: 10.1016/0092-8674(94)90302-6
- Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, et al. (2004) FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp Cell Res 297: 152–164. doi: 10.1016/j.yexcr.2004.03.011
- Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, et al. (2005) Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci 118: 5089–5100.
- Atsumi T, Miwa Y, Kimata K, Ikawa Y (1990) A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ Dev 30: 109–116. doi: 10.1016/0922-3371(90)90079-c
- Hoogendam J, Parlevliet E, Miclea R, Lowik CW, Wit JM, et al. (2006) Novel early target genes of parathyroid hormone-related peptide in chondrocytes. Endocrinology 147: 3141–3152. doi: 10.1210/en.2006-0075.
- Matsushita M, Kitoh H, Ohkawara B, Mishima K, Kaneko H, Ito M, et al. (2013) La meclozina facilita la proliferación y diferenciación de condrocitos al atenuar la señalización de FGFR3 anormalmente activada en la acondroplasia. PLoS ONE 8(12): e81569. https://doi.org/10.1371/journal.pone.0081569.
-
P.D. Sponseller, M.C. Ain. The skeletal dysplasias. Lovell & Winter’s pediatric orthopaedics, pp. 206-246