INFORMACIÓN BÁSICA: Síndrome de Sturge-Weber (SSW) o Angiomatosis encefalotrigeminal (ORPHA:3205/CIE-10:Q85.8/OMIM:185300).- El SSW (denominada la cuarta facomatosis) es un síndrome congénito Es esporádica y de etiología desconocida.
El SSW, es el más frecuente de los síndromes neurocutáneos, perteneciente a las denominadas Facomatosis. Se trata de un trastorno congénito, esporádico y de etiología desconocida, caracterizado por una malformación vascular cutánea (facial uni o bilateral), que afecta a las leptomeninges, al cerebro ipsilateral y ojos, caracterizadas por una mancha color vino de oporto en el área facial del nervio trigémino, malformación vascular de las leptomeninges, epilepsia, retraso mental, déficit neurológico, compromiso ocular, glaucoma y aumento de la vascularización de la retina. El diagnóstico precoz es primordial, por los estudios, interconsultas y terapéuticas a realizar; éste debe sospecharse aunque la clínica dermatológica no sea muy manifiesta.
Prevalencia.- Afecta ambos sexos por igual, no presenta carácter hereditario, pero se han descripto casos familiares. Tiene una incidencia de 1/50 000 a 1/230 000 personas.
Etiología.- La etiología de SSW está probablemente asociada con el mosaicismo somático. Huq et al informaron evidencia de mosaicismo somático en 4 pacientes con SSW. Dos tenían biopsia de piel de las manchas en vino de oporto, y los otros 2 tenían angiomas leptomeníngeos de hemisferectomía. Se observó la inversión del brazo cromosómico 4q y la trisomía 10 en un paciente cada uno. Shirley et al identificaron una mutación activadora somática c.548G-> A en GNAQ (en el cromosoma 9q21) en muestras de tejidos afectados, en 23 de los 26 participantes del estudio con SSW.
Anteriormente, la mutación somática recurrente c.548G4A (p.R183Q) en el gen G-α q (GNAQ) se identificó como causante en SSW y pacientes con tinción de vino de Oporto no sindrómico usando genoma completo secuenciación Mitsuko Nakashima et al investigaron mutaciones somáticas en GNAQ mediante secuenciación de próxima generación. Primero secuenciaciaron amplicones dirigidos de 15 muestras emparejadas de sangre-cerebro en SSW esporádica e identificaron la mutación somática recurrente en c.548G4A en el 80% de los pacientes (12 de 15). El porcentaje de alelos mutantes en los tejidos cerebrales de estos 12 pacientes varió de 3.6 a 8.9%. No encontraron otras mutaciones somáticas en ninguno de los siete exones GNAQ en los tres pacientes restantes sin c.548G4A. Estos hallazgos sugieren que la mutación GNAQ somática recurrente c.548G4A es el principal factor genético determinante para SSW e implica que otro (s) gen (es) candidato (s) mutado (s) pueden existir en SSW.
Se ha informado que los vasos corticales malformados en SSW están inervados sólo por las fibras nerviosas simpáticas noradrenérgicas y también se ha observado una mayor expresión de endotelina-1 en los vasos intracraneales malformados. Estos hallazgos pueden sugerir una mayor vasoconstricción en estos vasos sanguíneos anormales, ya que la endotelina-1 es un péptido asociado con la vasoconstricción.
La fibronectina es una molécula importante en la regulación de la angiogénesis, el mantenimiento de la barrera hematoencefálica y la estructura y función de los vasos sanguíneos, así como las respuestas de los tejidos cerebrales a las convulsiones. Comi et al informaron que, en pacientes con SSW, se observó una disminución de la expresión de fibronectina en los vasos sanguíneos leptomeníngeos, mientras que se observó una mayor expresión en los vasos parenquimatosos. La circunferencia de los vasos sanguíneos leptomeníngeos se redujo, mientras que la densidad de los vasos sanguíneos aumentó en SSW.
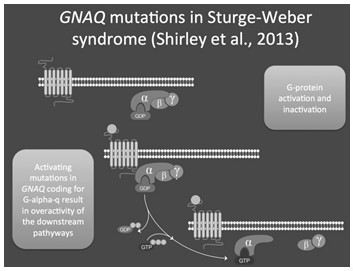
En general, en SSW, una mutación somática activante en el gen GNAQ (p.Arg183Gln), visto en la mayoría de los casos en el cromosoma 9 (en 9q21.2), parece causar alteraciones en la regulación de la estructura y la función de los vasos sanguíneos, inervación de los vasos sanguíneos y expresión de matriz extracelular y moléculas vasoactivas (Figura 14).
Patogenia.- El SSW es un síndrome no hereditario de etiología desconocida. Se piensa que está causado por mutaciones somáticas en el gen GNAQ 1 que afectan al primordio neural anterior antes de la migración de la cresta neural cefálica. Aunque en algunos pacientes se ha demostrado una expresión incrementada del gen de la fibronectina en fibroblastos obtenidos de tejidos lesionados, atribuyéndole un papel en la patogénesis.
Shirley et al., realizaron la secuenciación del genoma completo en tejido afectado o no afectado de tres individuos con SSW. En los tres individuos, identificaron la misma mutación c.548G-> A en GNAQ que da como resultado una sustitución de aminoácidos p.Arg183Gln. En una muestra más grande de 26 pacientes con SSW, identificaron la misma mutación en el 88% de los individuos. Asimismo, la mutación se encontró en tejido de biopsia afectado en 6/7 pacientes con manchas aisladas de vino de Oporto. En pacientes en los que estaba disponible la combinación de tejido afectado y no afectado, se pudo demostrar que la mutación era somática.
El gen GNAQ codifica para G-alfa-q, una subunidad de proteína G que participa en la señalización intracelular aguas abajo de proteínas transmembrana. Específicamente, GNAQ está implicado en la señalización dentro de la MAPK y ruta de la fosfolipasa C. Muchas de las señales celulares implicadas en el crecimiento celular operan a través de estas vías. Utilizando un ensayo informador para la vía MAPK, los autores pudieron demostrar que la mutación GNAQ dio como resultado una activación de la señalización aguas abajo. Esto sugiere que la mutación GNAQ produce una hiperactividad de esta vía. Se han notificado otras mutaciones somáticas activadoras en GNAQ en el melanoma uveal, una forma rara de cáncer de ojo. Las mutaciones subyacentes, sin embargo, conducen a una activación más fuerte de la vía MAPK. Esto sugiere que el grado de activación puede influir en el fenotipo resultante de la mutación somática.
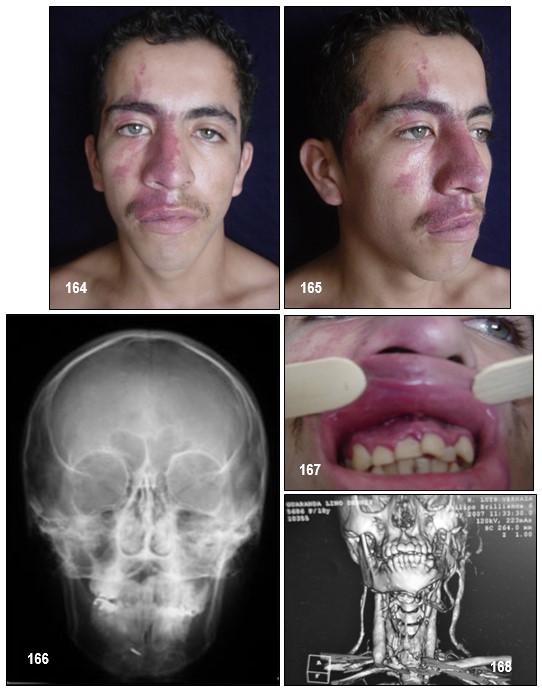
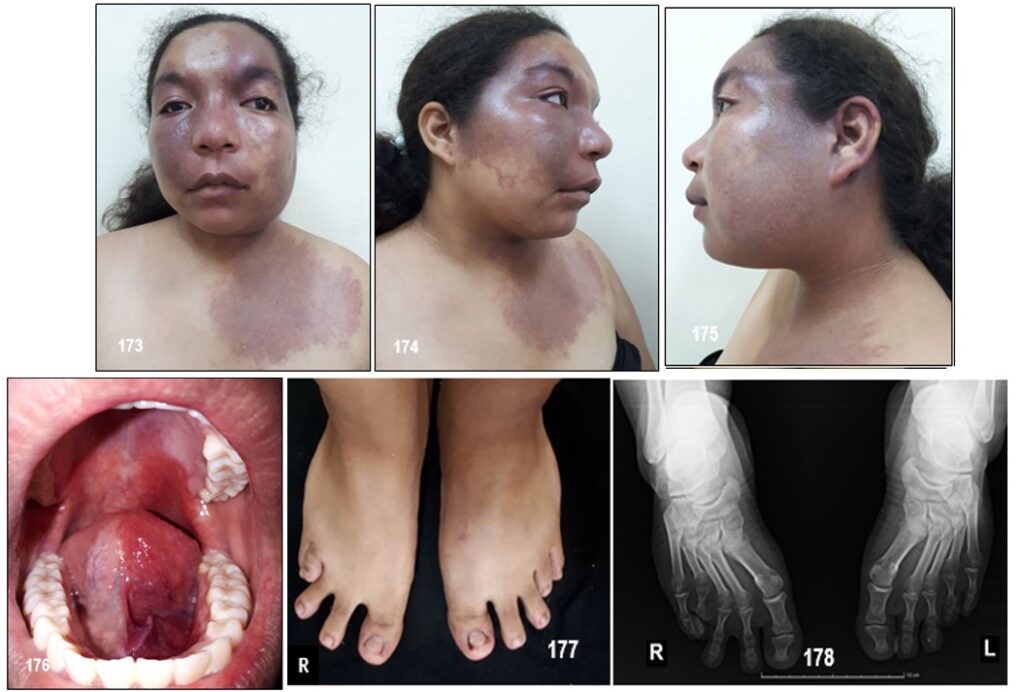
Manifestaciones clínicas.- Como en otras facomatosis, las lesiones se localizan en la piel, los ojos y el sistema nervioso. Las alteraciones fundamentales de la enfermedad son: angioma plano facial, epilepsia y calcificaciones cerebrales. Las mucosas de la boca resultan afectadas en un tercio de los enfermos. Las características clínicas neurológicas incluyen epilepsia, déficits neurológicos y problemas cognitivos. Las crisis epilépticas ocurren en el 75% de los pacientes, suelen ser el síntoma de presentación después de las manifestaciones cutáneas. Los déficits neurológicos focales van a depender de la localización de la lesión angiomatosa intracraneal, así en los casos que se afecta el territorio occipital presentara alteraciones campimétricas.
También pueden presentarse con hemiparesia.
Las cefaleas y migrañas también son frecuentes en los pacientes con SSW (Lissotto et al; 2004, Taddeucci et al; 2005). Casi un 83% de los pacientes presentan problemas cognitivos (Sujansky E 1995). Dos tercios presentan retraso psicomotor en la infancia y casi un 50% presentan un retraso mental significativo al final de la niñez. (Comi et al; 2006).
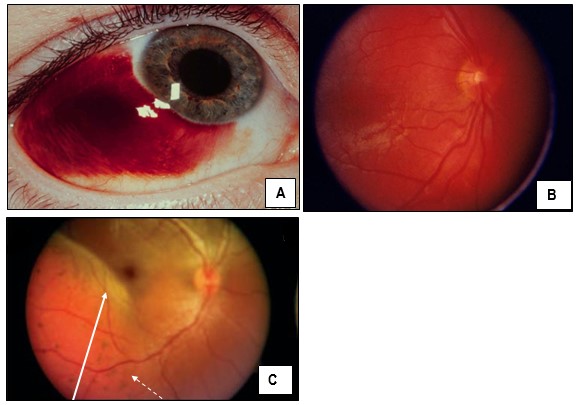
Con respecto a los síntomas oculares la anomalía de los vasos sanguíneos en el ojo, predispone al aumento de la presión intraocular y al desarrollo de glaucoma en un 60% de los casos. El angioma ocular aparece en el 30 % de los casos y afecta las coroides y la esclerótica ocular, y es ipsilateral al angioma cutáneo.
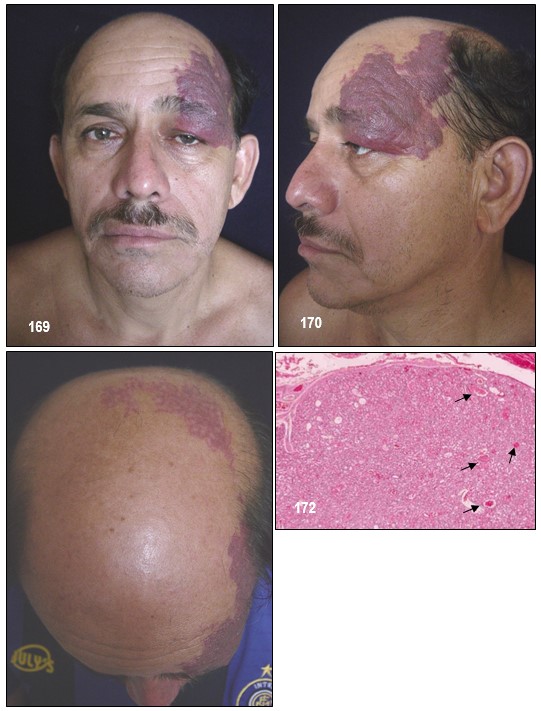
La malformación vascular capilar asociada, llamada «mancha en vino de oporto», compromete el área facial inervada por la primera rama sensitiva del nervio trigémino, particularmente el párpado superior y la zona supraorbitaria. Puede tomar las ramas maxilar y mandibular del mencionado nervio, con sobrecrecimiento óseo o
de las partes blandas de la zona. Este angioma cutáneo facial, el nevus en vino de oporto, presente en el nacimiento, frecuentemente en el territorio cutáneo inervado por primera rama del trigémino.
Únicamente entre el 10-20% de los pacientes que presentan estos hallazgos cutáneos presentan afectación del sistema nervioso central. El angioma leptomeníngeo suele ser unilateral pero en un 15% de los casos es bilateral. También puede presentarse las características neurológicas y neuroradiológicas típicas sin afectación cutánea, aunque esto es menos frecuente (Comi et al; 2003). En la evolución, la lesión tiende a oscurecerse y a adquirir zonas nodulares que imprimen a la lesión un aspecto desagradable como lo he podido observar. Puede estar ausente en la forma «frustra» del síndrome. El 37% de los pacientes presenta lesiones bilaterales, las cuales pueden estar asociadas a compromiso intracraneal unilateral o bilateral y el 36% puede tener malformaciones vasculares en las extremidades o el tronco. Se han descripto casos de localizaciones atípicas, o sin malformación facial evidente, como cuero cabelludo o mucosa oral, fuera del área trigeminal, asociados a anomalía leptomeníngea.
Ante la sospecha diagnóstica es oportuno realizar interconsultas con neurología y oftalmología; asimismo, solicitar estudios por imágenes para descartar patologías asociadas.
Las manifestaciones neurológicas se relacionan con las malformaciones vasculares que afectan la piamadre o angiomas leptomeningeos (angioma pial), suelen ser ipsolaterales a la mácula cutánea y, frecuentemente, se asocian calcificaciones ipsolaterales, hemiatrofia cerebral progresiva y otras anormalidades capilares y venosas. También la alteración vascular pial puede aparecer aislada en el 13% de los pacientes, sin el angioma de la coroides ni facial.
El curso clínico de los portadores del SSW guarda una relación estrecha con las malformaciones neurológicas y los cambios neurorradiológicos, pero no con el tamaño de la lesión vascular facial; todo el hemisferio cerebral puede estar afectado.
La manifestación neurológica más constante es la presencia de convulsiones, que afectan al 75- 90% de los pacientes. En el 45%, las crisis epilépticas se inician antes del año de vida, con un pico entre los 3 y 6 meses de edad. Sólo el 7% de los pacientes inicia sus crisis después de los 5 años. Al principio, son comunes las crisis parciales motoras o las crisis tónicoclónicas generalizadas. Con menos frecuencia ocurren los espasmos infantiles, las crisis mioclónicas y atónicas.
Es usual la presencia de crisis frecuentes y prolongadas de difícil control farmacológico, que se logra controlar en el 47% de los pacientes. El inicio de las crisis por debajo de los 2 años hace más probable su refractariedad; algunos autores refieren que las crisis epilépticas son más frecuentes e intensas cuando existe afectación bihemisférica.
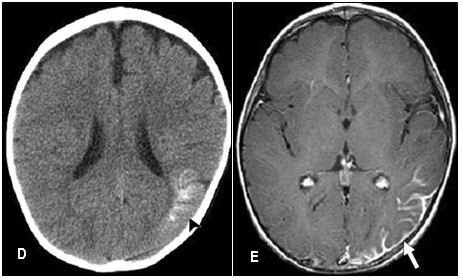
Puede haber déficit neurológico focal: como defecto motor hemiparético. (30-40%), que simula una parálisis de Todd. Persiste durante mayor tiempo, puede hacerse permanente. 31% de los pacientes presenta hemiatrofia de los miembros contralaterales al angioma pial. Puede presentarse también hemianopsia homónima, cuadrantanopsia homónima y ceguera.
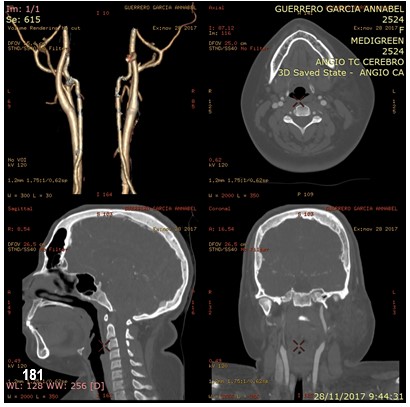
La RM craneal con gadolinio, permite visualizar las alteraciones de los vasos intracraneales en los pacientes con SSW. La angiografía demuestra disminución o ausencia de venas superficiales corticales en el área de la lesión y aumento de las colaterales.
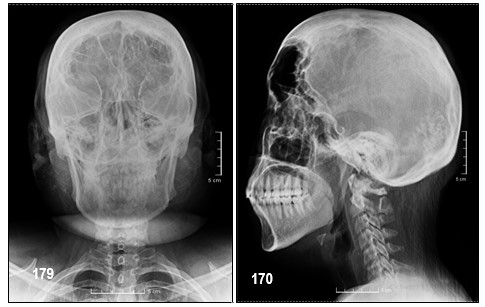
El retraso mental afecta al 50-70% de los pacientes con posterioridad al inicio de las crisis epilépticas; el 2,5% desarrolla retraso mental grave. Sólo el 8% de aquellos con lesiones bilaterales son intelectualmente normales. Las imágenes características de tomografía axial computada (TAC) cerebral o RMN, con gadolinio o sin él, pueden sugerir el diagnóstico aun antes del comienzo de las manifestaciones clínicas del SNC. La RMN ofrece mejor detalle que la TAC. Se puede observar la malformación vascular leptomeníngea que resulta en isquemia en los tejidos adyacentes, gliosis, desmielinización, calcificaciones, hemiatrofia cerebral y atrofias focales; ausencia de venas corticales superficiales adyacentes a la malformación y aumento de tamaño del sistema venoso profundo y del plexo coroideo ipsolaterales. Las calcificaciones con patrón de doble contorno que siguen las circunvoluciones cerebrales (en «vías de tren») se observan con la TAC, desde los primeros meses de vida y, con la radiografía de cráneo, desde los 7 años.
- – Calcificaciones corticales giriformes adyacentes al angioma leptomeníngeo.
- – Puede observarse en TAC. Generalmente se encuentran en región occipital.
- – Angioma pial: Puede ser enmascarado por las calcificaciones. Visible en TAC.
- – RM: suele mostrar atrofia y calcificaciones.
- – RM con gadolinio: Estudio más fidedigno para valorar la extensión del angioma pial.
Los estudios Imagenológicos evalúan:
- Extensión de la malformación angiomatosa leptomeníngea.
- Grado de atrofia del parénquima.
- Posibles cambios isquémicos que afectan sustancia gris y blanca.
- Presencia y extensión de calcificaciones corticales.
- Prominencia de plexos coroides.
- Anomalías venosas del parénquima.
El EEG típico en estos pacientes es asimétrico, con actividad del hemisferio afectado reducida en voltaje y enlentecida. La afectación ocular es ipsolateral a la MV; se constata glaucoma, buftalmos, malformaciones vasculares de la conjuntiva, esclerótica, coroides y retina. Con menor frecuencia, heterocromía del iris, atrofia óptica, opacificación corneal y desprendimiento de retina. Estos hallazgos no fueron observados en nuestro paciente.
En el 25% de los pacientes pueden encontrarse lesiones asociadas en la boca, principalmente alteraciones vasculares de los labios y la mucosa oral, sobrecrecimiento óseo o de partes blandas y engrosamiento gingival, como en nuestro paciente.
El diagnóstico es clínico, al observar una malformación vascular facial en el territorio del trigémino y alteraciones en SNC. Además de las nuevas técnicas de RM que pueden ayudar en el diagnóstico temprano, los estudios con SPECT y PET indican que la reducción del flujo sanguíneo en el cerebro, junto con los cambios en la circulación causados por las crisis prolongadas, puede empeorar el daño neurológico en esta condición.
Diagnóstico diferencial.- Dentro de los diagnósticos diferenciales debemos considerar: anomalías vasculares aisladas o asociadas a síndromes: facomatosis pigmento-vascular; Síndromes de: Parkes-Weber, Rubinstein-Taybi, síndrome PHACES, defectos cardiacos y coartación aortica, anomalías oculares (microftalmia, atrofia de nervio óptico), malformaciones en fosa posterior: Hipoplasia cerebelosa, malformación de Dandy-Walker, entre otros.
Tratamiento.- La terapéutica de la epilepsia, será farmacológica o quirúrgica (o incluirá ambas). Las convulsiones responden a terapéutica anticonvulsiva en el 50% de los casos. Se recomienda el empleo de agentes antitrombóticos (aspirina) en lesiones de tamaño considerable, pues disminuyen los episodios trombóticos y el deterioro progresivo.
La hemisferectomía está indicada sólo en pacientes con malformación vascular leptomeníngea extensa unilateral, con hemiparesia unilateral y convulsiones intratables. Vázquez y col., en su trabajo sobre hemisferectomías y hemi-hemisferectomías, manifiestan que, para el manejo de las epilepsias refractarias al tratamiento farmacológico, dichos procedimientos ofrecen un alto índice de resultados positivos con baja morbimortalidad.
El tratamiento del glaucoma es, en general, arduo y los pacientes requieren cirugía.
La malformación vascular facial en general representa un problema cosmético. La terapia con láser de colorante pulsado es la mejor alternativa de tratamiento estético, con bajo riesgo y dolor variable. Se pueden también utilizar maquillajes cubritivos.
La estimulación precoz se destaca para mejorar el retraso mental y los déficit motores.
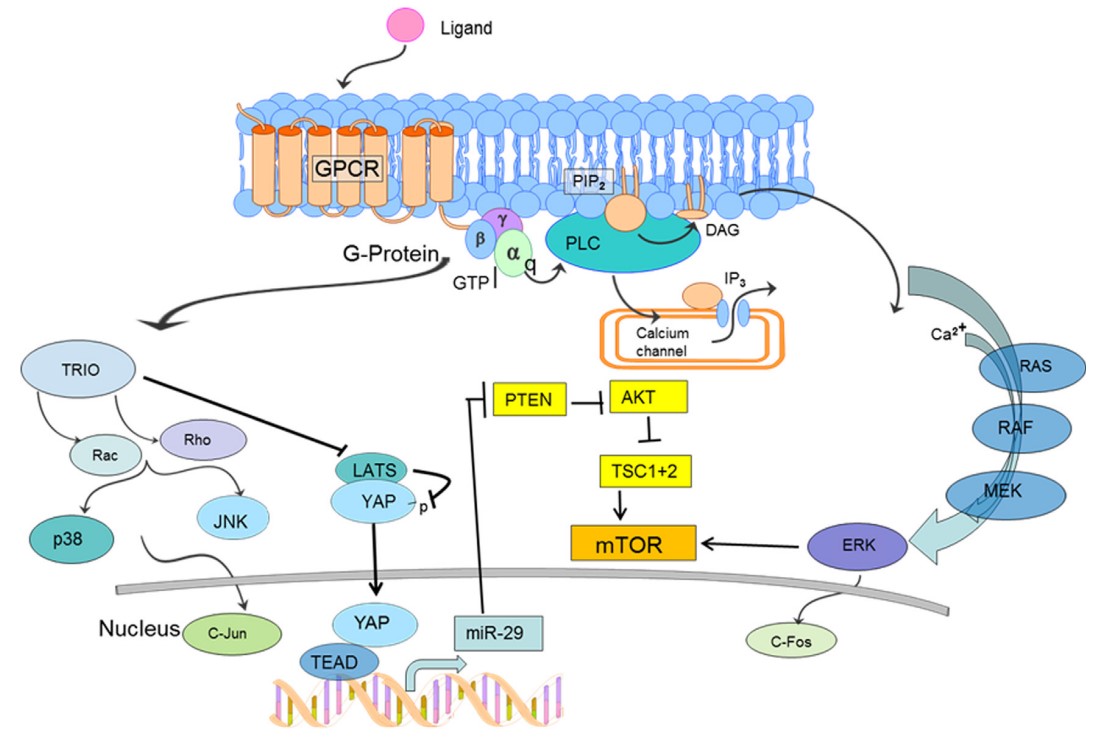
Figura 1. Tratamiento potencial mediante la inhibición de la activación constitutiva de la vía Hippo-YAP y Ras-Raf-MEK-ERK-mTOR mutación somática en GNAQ provoca una alteración de la autohidrólisis de Gαq con una sobreactivación vías descendentes, incluida una mayor actividad de mTOR. Actualmente se está estudiando el tratamiento con inhibidores de mTOR. mTOR, diana de la rapamicina. (La versión en color de la figura está disponible en línea). Fuente: Comi A. Current therapeutic options in sturge-Weber syndrome. Semin Pediatr Neurol. 2015;22(4):295-301
Comentario de los Editores: EL SSW se presenta desde el nacimiento, y cuya malformación vascular (angioma plano), se distribuye en un costado de la cara y del cráneo (pacientes que presentamos), y que corresponden a las ramas vasculares 1ra y 2da. En un tercio puede abarcar también la rama 3ra del trigémino, o ser bilateral. Con el tiempo llega a ocasionar hipertrofia de las partes blandas y a mostrar en su superficie elevaciones vasculares papulosas irregulares. El compromiso de la rama V1 y V2 da una sólida presunción de acompañarse de angiomatosis leptomeningea. La cara pronostica el riesgo del cerebro.
Bibliografía
- Lisotto C, Mainardi F, Maggioni F, Zanchin G. (2004) Headache in Sturge-Weber syndrome: a case report and review of the literature. Cephalalgia 24:1001–1004.
- Sujansky E, Conradi S (1995). Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet 57:35–45.
- Taddeucci G, Bonuccelli A, Polacco P (2005). Migraine-like attacks in child with Sturge-Weber syndrome without facial nevus. Pediatr Neurol; 32:131–133.
- Comi AM, Fischer R, Kossoff EH. (2003) .Encephalofacial angiomatosis sparing the occipital lobe and without facial nevus: on the spectrum of Sturge-Weber syndrome variants? J Child Neurol 2003; 18:35–38
- Comi AM. (2006). Advances in Sturge-Weber syndrome Curr Opin Neurol. 19(2):124-8.
- Vázquez C, de Jesús Barrios L, Bartuluchi M, Medina C, et al. Hemisferectomías y Hemisferectomías: nuestra experiencia acerca de 49 casos. Rev Argent Neuroc 2008; 22(5):131-133.
- Sturge WA.: A case of partial epilepsy, apparently due to a lesion of one of the vasomotor centres of the brain. Trans Clin Joc. (1879); 12:162-7.
- Weber F.P.: Right-sided hemihypertrophy resulting from right sided congenital spastic hemiplegia, with a morbid condition of the left side of the brain revealed by radiograms. J Neuro Psychopathol. (1922);3:134-9.
- Dimitri V.: Tumor cerebral congénito (angioma cavernosum). Rev Ass Med Argent. (1923); 36: 63.
- Krabbe K. H.: Facial and meningeal angiomatosis associated with calcification of the brain cortex. A clínicasl and an anatomopathologic contribution. Archives of Neurology and Psychiatry. (1934); 32:737-55
- Rodríguez A.C.: Síndromes neurocutáneos de anomalías vasculares. Rev Neurol. (1996);24(133):1072-84
- Rodríguez E. y Unamono P.: El síndrome de Sturge-Weber. Med Clin. (1993); 101:18-
- Fernandez Concepción, Gomez García et al., Síndrome de Sturge Weber. Revisión. Rev CubanaPediatr 1999; 71 (3):153-9.
- Convusions and homonymous hemianopsia as initial manifestations of Sturge-Weber syndrome in a 64-year-old male. García-Estévez DA. Neurologia. 2013 Mar 2. doi:pii: S0213-4853(13)00010-8. 10.1016/j.nrl.2013.01.004. [Epub ahead of print] English, Spanish.
- Huq AH, Chugani DC, Hukku B. Evidencia de mosaicismo somático en el síndrome de Sturge-Weber. Neurología. 2002 Sep. 10. 59 (5): 780-2.
- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Síndrome de Sturge-Weber y manchas de vino de puerto causadas por mutación somática en GNAQ. N Engl J Med . 2013 23 de mayo. 368 (21): 1971-9.