Nota de los Editores.- Esta enfermedad no es rara en Europa. No pertenece a la nomenclatura de enfermedades raras de Orphanet.
INFORMACION BÁSICA.- Síndrome de Klinefelter (SK) (OMIM/ORPHA – /CIE-10:Q98.0).- El SK es una anomalía cromosómica que afecta solamente a los hombres y ocasiona, principalmente, hipogonadismo. Se basa en una alteración genética que se desarrolla por la separación incorrecta de los cromosomas homólogos durante las meiosis que dan lugar a los gametos de uno de los progenitores, aunque también puede darse en las primeras divisiones del cigoto.
El sexo de las personas, como bien podríamos saber, está determinado por los cromosomas X e Y. Los varones tienen los cromosomas sexuales XY (46, XY) y las mujeres tienen los cromosomas sexuales XX (46, XX). En el SK, el hombre cuenta, como mínimo, con un cromosoma X extra, dando lugar en el 75% de los casos a un cariotipo (47, XXY). No obstante, un 20% de los casos, aproximadamente, son mosaicos cromosómicos, con variantes como (48, XXXY), (48, XXYY), y (49, XXXXY) en el 5% de los casos. Como regla general, los cromosomas X adicionales generan un correspondiente fenotipo más anormal (aunque los cromosomas X adicionales son inactivos), con un grado mayor de dismorfismo, desarrollo sexual más defectuoso y deterioro mental más grave. Una observación inesperada en los pacientes con el mosaisismo 49, XXXXY (y en sus contrapartidas femeninas, con cariotipos, 49, XXXXX) es que el fenotipo resulta similar en muchos aspectos del síndrome de Down.
Se cree que Carlos II de España, el último rey de la casa de Austria, sufrió este síndrome debido, fundamentalmente, a las sucesivas uniones endogámicas de sus antepasados y la degeneración familiar por falta de sangre nueva.
El SK o disgenesia gonadal., o sea, de los túbulos seminíferos se considera la anomalía cromosómica más común en los humanos, presentándose con una incidencia de 1 en 500 en los recién nacidos vivos varones. Los afectados presentan un cromosoma X supernumerario, lo que conduce a un fallo testicular primario cuyas consecuencias primarias son infertilidad e hipoandrogenismo. A pesar de la relativa frecuencia del padecimiento en recién nacidos vivos, se estima que la mitad de los productos (47, XXY) se abortan de manera espontánea. El SK es considerado la causa más frecuente en hombres de hipogonadismo. El SK Fue descrito en 1942 por Harry Klinefelter y colaboradores, que estudiaron nueve pacientes con: ginecomastia, testículos pequeños, azoospermia y elevada concentración de gonadotropinas (LH y FSH). Ellos sugirieron que el defecto primario estaba en las células de Sertoli y propusieron que, además, en estos pacientes había una deficiencia en una hormona testicular que regulaba la concentración de gonadotropinas hipofisiarias, a la que llamaron hormona X o inhibina.
En 1956 se demostró la presencia del corpúsculo de Barr en pacientes con SK y tres años más tarde se identifica que el cariotipo de un sujeto con la enfermedad era (47, XXY). De esta manera se estableció que la presencia de un cromosoma X extra era el factor etiológico fundamental para desarrollar las características de dicho síndrome. A pesar de la generalidad extendida de que los varones que presentan SK van a desarrollar un determinado fenotipo, muchos de ellos no lo hacen, pudiendo llevar una vida normal. En estos casos, el síndrome se hará evidente en la edad adulta, cuando el individuo acuda al especialista por problemas de fertilidad, siendo entonces cuando se detecta el cromosoma extra y se diagnostica la causa de la esterilidad. Debido a estos casos, muchos médicos e investigadores están empezando a dejar en desuso el término «síndrome de Klinefelter», usando en su lugar la descripción de «varones XXY».
Causa: El cromosomaX adicional en los pacientes con SK a menudo es adquirido por un error en la disyuncióndurante la meiosis I (gametogénesis). El error en el proceso de disyunción (separación de cromosomas durante la división celular) se da cuando cromosomas homólogos (en este caso, los cromosomas sexuales X e Y) fallan al separarse, originando gametos (masculinos o femeninos) con 24 cromosomas, debido a dicho cromosoma adicional.
Según las estadísticas, un 56% de individuos (47, XXY) tienen como causa del síndrome la no disyunción materna durante la meiosis I o la meiosis II. En esta última, las cromátidas hermanas de ambos cromosomas X no se separan correctamente, produciéndose un óvulo XX que, al ser fertilizado por el esperma, portador del cromosoma Y, da lugar a un embrión XXY.
El 44% restante de los casos tienen su origen en errores durante la meiosis I paterna, tal y como ocurre en las trisomías autosómicas. Estos fallos durante meiosis suelen ser aislados, si bien se ha comprobado que la probabilidad de errores durante la meiosis I materna aumenta con la edad ésta. La anomalía cromosómica puede originarse también por un error durante las divisiones mitóticas del cigoto, produciendo así los casos de mosaicismo.
En mamíferos con más de un cromosoma X (en el caso de humanos, mujeres), se da la inactivación de uno de los dos cromosomas, de modo que se equipare la carga génica con el hombre. Esto también ocurre en los varones XXY, aunque hay cierta evidencia que sugiere que algunos genes localizados en las regiones pseudoautosomales de sus cromosomas X presentan correspondencia con su cromosoma Y, siendo capaces de expresarse. Los estudios en sujetos prepuberales (47, XXY) no muestran deficiencias en las concentraciones de LH, FSH o testosterona, comparados con sujetos prepúberes (46, XY) y la respuesta a la gonadoliberina (LHRH, hormona hipotalámica liberadora de gonadotropinas) es normal en ambos grupos. Sin embargo, entre los 12 y 14 años de edad en los sujetos (47 XXY) las concentraciones de gonadotropinas se incrementan y la testosterona permanece en límites inferiores para la edad.
En biopsias realizadas a niños con el síndrome se ha observado sólo disminución en el número de células germinales. No obstante después de la pubertad se aprecia hialinización y fibrosis de los túbulos seminíferos, que son los cambios histológicos característicos del síndrome y que originan disminución en el volumen testicular y aumento de su consistencia. Además, se observa ausencia de células germinales, hiperplasia y agregación de las células de Leydig, como repuesta a la hiperestimulación por la LH. Las alteraciones histológicas se hacen más frecuentes con la edad.
La pérdida de túbulos seminíferos y células de Sertoli produce una disminución en las cifras de inhibina B, el factor regulador de FSH, y de AMH u hormona antimülleriana, lo que disminuye la retroalimentación negativa sobre la FSH, aumentando ésta. La ausencia de espermatogénesises secundaria a la presencia de cromosomas supernumerarios, que se mantienen activos durante la gametogénesis.
Característica clínicas: A continuación, se listan las características más comunes en los varones XXY. No obstante, no todas ellas aparecen en un mismo individuo.
- – En edad temprana (en la niñez), cuando el varón es XXY es un bebé, suele presentar una musculatura menos desarrollada y fuerza reducida.
- – Suelen gatear y comenzar a andar de forma más torpe y tardía que los demás niños.
- – Talla elevada en la edad adulta. Suelen tener mayor estatura que sus padres y hermanos, y mayor altura en comparación con la altura media de un hombre. Se caracterizan por tener las extremedidades muy largas en relación al tamaño del cuerpo.
- – Mayor propensión a padecer enfermedades autoinmunes, cáncer de pecho, alteraciones venéreas, osteoporosis y algunas alteraciones dentarias.
- – Mayor acumulación de grasa subcutánea y mayor tendencia al sobrepeso. Los varones XXY poseen un cuerpo más redondeado, en forma de pera, característico de la mujer. Esto se debe a que desarrollan caracteres femeninos, siendo uno de ellos el poseer caderas más anchas o acumular grasa en zonas características de la mujer.
- – Dismorfia facial discreta.
- – A veces, criptorquidia, micropene, escroto hipoplásico o malformaciones de genitales, atrofia testicular (1-2cm3) (n: 3,5 a 4cm3).
- – Esterilidad por azoospermia.
- – Ginecomastia uni o bilateral. Se caracteriza por el desarrollo de pechos en el hombre (tejido mamario agrandado).
- – Escasez de vello en la cara y en todo el cuerpo. Es consecuencia directa de la baja concentración de testosterona.
- – Vello pubiano disminuido, o siguiendo un claro patrón femenino.
- – Gonadotrofinas elevadas en la pubertad (LH y FSH). Hipogonadismo hipergonadotrófico.
- – Disminución de la libido sexual en la edad adulta.
- – Retraso en el área del lenguaje, lectura y comprensión. Los niños XXY por lo general aprenden a hablar mucho más tarde que los otros niños, y pueden tener ciertas dificultades para leer y escribir. Muchos de ellos suelen tener algún grado de dificultad con el lenguaje de por vida. Sin embargo, los varones XXY presentan un coeficiente intelectual normal.
- – Lentitud, apatía y retraso en el desarrollo emocional.
- – Trastornos emocionales, ansiedad, depresión, etc.
- – Falta de autoestima, debida en la mayoría de los casos a los caracteres femeninos perceptibles por el varón (ginecomastia, etc.).
- Diagnóstico: El SK puede presentarse como:
- – Niño con retraso leve en las adquisiciones y comportamiento inmaduro.
- – Adolescente con testículos pequeños y de menor consistencia.
- – Adulto con hábito eunucoide, ginecomastia y escaso desarrollo muscular.
- – Adulto con infertilidad.
Sin embargo en los últimos años muchos casos se diagnostican prenatalmente. El diagnóstico definitivo lo dará el estudio de los cromosomas.
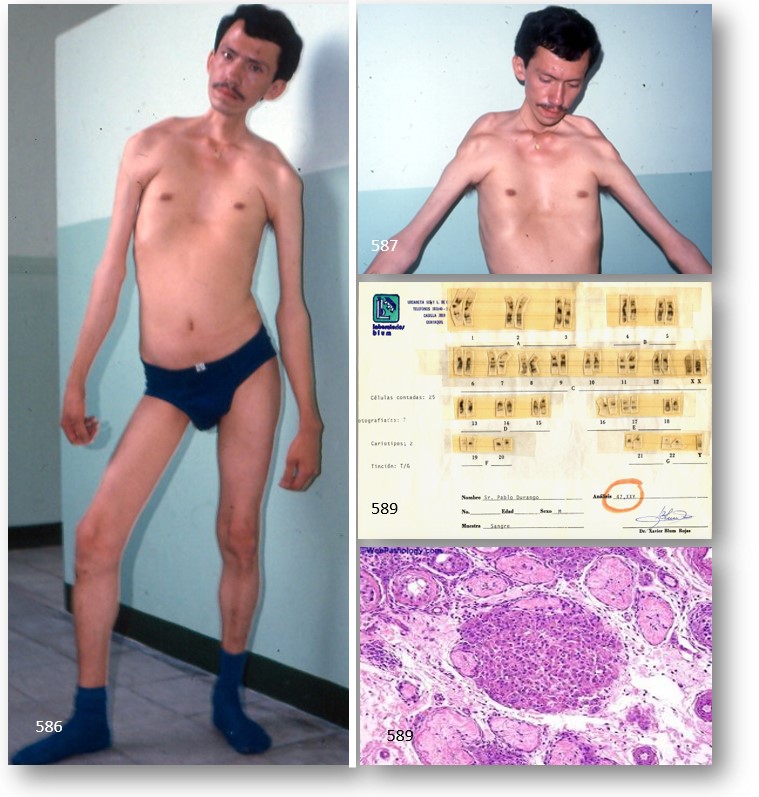
Ante la sospecha de un posible SK, se procede a la realización de un cariotipo (imagen 3). Para ello, se toma una muestra de sangre, de la cual se separan los leucocitos o glóbulos blancos, se incuban y se hace un estudio de los cromosomas para detectar anomalías, como en este caso, la presencia de un cromosoma X extra. Deben contarse un número adecuado de células para detectar mosaicismo. Los niveles de testosterona en el plasma están bajos con valores normales o elevados de LH y FSH (hipogonadismo hipergonadotrófico).
Otra forma de diagnosticar un Klinefelter de forma prenatal es por amniocentesis o por la muestra del villus coriónico. Ambos son test en los que se extrae tejido del feto para examinar su ADN en busca de anomalías genéticas. En 2002, se hizo un estudio sobre la tasa de interrupción de embarazos como consecuencia de un diagnóstico genético. En él se indica que el 58% de las embarazadas en EEUU decidieron interrumpir el embarazo debido a un diagnóstico positivo de Klinefelter.
Diagnóstico diferencial: Hábitos similares se aprecian en la homocistinuria y en el síndrome de Marfan; otras causas de hipogonadismo. No debe confundirse con síndrome del XYYo síndrome del triple X).
Tratamiento: Afortunadamente, la mayor parte de estos síntomas se pueden tratar, de modo que el varón XXY pueda evitar los numerosos problemas psicológicos derivados de las características de un Klinefelter. Por ejemplo, con una cirugía se puede reducir el tamaño de los pechos. A su vez, una hormonoterapia de sustitución de la testosterona, comenzada desde la pubertad, puede promover el crecimiento de vello en la cara, así como un cuerpo más musculoso.
Otras manifestaciones más preocupantes en la edad adulta, como la infertilidad, permanecían sin solución. Sin embargo, en el año 2010 se habían registrado hasta 100 embarazos satisfactorios llevados a cabo por fecundación in vitro utilizando material espermático extraído quirúrgicamente de hombres XXY.
Seguimiento: Se debe tener en cuenta que no todos los varones con cariotipo 47, XXY manifestarán todas estas características.
- – Sistema musculoesquelético: La masa muscular es poco desarrollada, por lo que el cansancio es más fácil. Pueden presentar una displasia leve a nivel de la articulación del codo, y clinodactilia del 5º dedo de las manos. La osteoporosis aparece sobretodo en los individuos que no reciben testosterona. Los adolescentes presentan escoliosis con más frecuencia que la población general. Los individuos con cariotipo 48, XXXY pueden tener talla baja y sinóstosis radio-cubital.
- – Desarrollo sexual: La pubertad aparece a una edad normal, pero los testículos no se desarrollan y permanecen pequeños. Los caracteres sexuales secundarios se desarrollan poco. El vello corporal es escaso y la distribución puede ser ginecoide. El tejido celular subcutáneo también puede adoptar una distribución femenina sobre todo a nivel de las caderas, y pueden presentar ginecomastia. La actividad sexual generalmente es normal o levemente deprimida. Debido al exceso de gonadotropina se produce de forma progresiva una hialinización y fibrosis de los túbulos seminíferos, con una inadecuada producción de testosterona y azoospermia en la mayoría de casos, requiriendo por ello tratamiento con testosterona a largo plazo. La mayoría de ellos son infértiles. Ocasionalmente pueden presentar criptorquídia e hipospadias.
- – Capacidad intelectual: El coeficiente intelectual de estos individuos es, ligera pero significativamente, inferior que el de los varones con cromosomas normales. Dos tercios tienen problemas de aprendizaje, especialmente dislexia. El lenguaje expresivo, la capacidad de procesamiento auditivo y la memoria auditiva son deficientes, lo cual conlleva una menor habilidad para leer y escribir.
- – Carácter: Los trastornos del comportamiento son frecuentes, especialmente inmadurez, inseguridad, timidez, y poca capacidad de juicio. Les cuesta relacionarse con individuos de su grupo de edad y pueden tener problemas de adaptación social. La depresión es frecuente en estos individuos.
- – Sistema nervioso: Puede aparecer ataxia. Entre un 20 y un 50% pueden tener un temblor intencional.
- – Sistema venoso: La enfermedad varicosa y las úlceras de extremidades inferiores pueden ser los primeros síntomas de los varones 47XXY.
- – Enfermedades autoinmunes: Existe un mayor riesgo de desarrollar enfermedades autoinmunes como diabetes, artritis reumatoide, tiroiditis y el lupus eritematoso
- – Neoplasias: Los varones XXY con ginecomastia tienen mayor riesgo de cáncer de mama.
Se ha descrito una mayor incidencia de tumores germinales extragonadales con afectación principalmente mediastínica.
La orientación sexual será generalmente heterosexual, pero puede haber disminución en la libido. Pacientes con 48XXXY y 49 XXXXY tienen síntomas más marcados y el retraso mental es común.
El riesgo de recurrencia, en el caso de una pareja con un hijo 47, XXY es del 1%. Este riesgo aumenta en mujeres por encima de los 40 años. Riesgo de incrementos de neoplasias de células germinales.
Addedum: Miositis por cuerpos de inclusión (IBM): La miositis por cuerpos de inclusion (Inclusion-body Myositis; IBM) fue descrita por primera vez en 1975 por Yunis y Samaha, que definieron una entidad clínicamente similar a una polimiositis crónica, caracterizada anatomopatológicamente por la presencia de inclusiones vacuolares con productos de degradación citoplasmáticos, así como cuerpos fibrilares en el núcleo y el citoplasma de las fibras musculares, acompañado de un infiltrado linfocítico. Ninguno de los hallazgos clínicos, neurofisiológicos o patológicos es específico de esta entidad, ya que estas inclusiones vacuolares se han descrito en otras miopatías distales y proximales, tanto esporádicas como familiares. Se ha considerado a la miositis por cuerpos de inclusion como una miopatía inflamatoria, junto a la polimiositis y la dermatomiositis, si bien numerosas observaciones la separan de este grupo, como por ejemplo la pobre respuesta a tratamientos inmunosupresores o la ausencia de infiltrado inflamatorio en algunos casos, especialmente en las formas de miopatía hereditaria. La miositis por cuerpos de inclusión está despertando un interés creciente a raíz de la identificación de cambios degenerativos en la fibra muscular, con la presencia de proteína amiloide anormalmente plegada y otros hallazgos patológicos que sugieren un proceso subyacente distinto al meramente inmunológico.
Patogenia.-Actualmente se consideran dos posibles hipótesis como causa de la enfermedad: un proceso degenerativo relacionado con depósitos amiloides y un fenómeno disinmune. Probablemente ambos mecanismos se encuentran interrelacionados.
Clínica.- La miositis por cuerpos de inclusión es la causa más frecuente de miopatía en personas mayores de 50 años, mostrando cierta predominancia masculina en las series descritas (3:1).
La forma clásica de la enfermedad suele comenzar entre los 50 y los 70 años de edad, acompañándose de debilidad muscular progresiva, proximal y distal, generalmente simétrica, si bien a veces aparece cierta selectividad por determinados grupos musculares. La evolución de la enfermedad suele conducir a una discapacidad grave. Los síntomas suelen aparecer simultáneamente en miembros superiores e inferiores, o bien hacerlo de forma secuencial. En las extremidades inferiores, suele afectar predominantemente al cuádriceps y glúteo mayor, y distalmente produce pie caído, con la consiguiente dificultad para la marcha, tropiezos y caídas frecuentes. En miembros superiores, la mayor afectación corresponde a los músculos flexores de los dedos, con menor participación del bíceps o el tríceps.
De forma característica, el flexor profundo de los dedos suele estar más afectado que el superficial. La alteración en miembros superiores conduce a un grado progresivo de discapacidad, con dificultad para la manipulación fina de objetos. El proceso suele acompañarse de atrofia, que suele ser proporcional al grado de debilidad. En ciertos casos se afecta la musculatura escapular, cervical o facial, siendo excepcional la debilidad de distribución bulbar, a veces acompañada de disfagia. La musculatura extraocular está respetada en todos los casos, pero en algunos pacientes puede haber afectación de la musculatura del tronco con compromiso de la función respiratoria. Algunos sujetos presentan mialgias transitorias, sobre todo al comienzo de la enfermedad.
Inicialmente los reflejos tendinosos están conservados, pero hasta en el 40% de los casos disminuyen con el transcurso de la enfermedad. Algunos casos se acompañan de polineuropatía difusa y sólo estos muestran ocasionalmente déficits sensitivos. Hasta un 18% de los pacientes muestra alteraciones del músculo cardíaco con miocardiopatía clínicamente definida, y en algunos casos se ha asociado un fenómeno de Raynaud.
Asociación de la IBM con otras enfermedades sistémicas.- La IBM se ha asociado con diabetes mellitus, neuropatía periférica sensitiva crónica, cirrosis, gota, celiaca del adulto, úlcera duodenal, tuberculosis pulmonar, síndrome de Klinefelter, parotiditis, alcoholismo y algunos tumores malignos, así como con otras enfermedades autoinmunes (Sjögren, lupus eritematoso, esclerodermia, sarcoidosis o trombocitopenia autoimmune).
Sin embargo, las investigaciones para detectar posibles autoanticuerpos involucrados en la IBM nunca han sido concluyentes, y los parámetros de inflamación como la velocidad de sedimentación (VSG) suelen ser normales o sólo levemente elevados, al igual que la cifra sanguínea de creatin kinasa (CK). Recientemente se han descrito asociaciones de esta entidad con mutaciones del gen de la transtiretina (amiloidosis), seropositividad para VIH y síndrome postpolio.
Hallazgos neurofisiológicos.- El electromiograma (EMG) suele anormal, pero no diagnóstico. Pueden aparecer hallazgos de denervación reciente (fibrilación y ondas positivas). Suele haber pequeños potenciales de unidad motora que reflejan una disminución del número de fibras activadas por unidad motora y que no es específico de un patron miopático. Puede haber evidencia de reinervación con unidades motoras prolongadas, pero generalmente menos frecuente de lo esperado para la cronicidad del proceso. En el electroneurograma (ENG) puede aparecer un patrón de polineuropatía desmielinizante que si es muy prominente puede plantear el diagnóstico diferencial con la polirradiculopatía inflamatoria desmielinizante crónica (CIDP), especialmente si hay aumento de proteínas en líquido cefalorraquídeo, bandas oligoclonales u otros marcadores de disinmunidad.
Conclusiones.- La IBM esporádica es la miopatía más frecuente en la edad adulta y una importante causa de discapacidad dado su carácter progresivo hacia un estado de incapacidad más o menos severo. En el momento actual no existe un tratamiento eficaz, si bien los nuevos hallazgos sobre la patogenia de este proceso pueden abrir nuevos caminos de investigación que conduzcan al desarrollo de estrategias terapéuticas. En este sentido son destacables las recientes aportaciones sobre el proceso degenerativo que tiene lugar en la fibra muscular y que asemeja al encontrado en el cerebro de pacientes con enfermedad de Alzheimer, en el que probablemente se vean implicados los sistemas proteícos del complejo ubiquitina-proteasoma, así como fenómenos de oxidación celular que conduzcan a una acumulación de productos anómalos con la consiguiente degeneración de la fibra muscular.
Bibliografía
- Sotos JF. Genetic disorders associated with overgrowth. Clin Pediatr 1997;36:39-49
- Smyth CM, Bremner WJ. Klinefelter syndrome. Arch Intern Med 1998;158:1309-1314
- Greenspan. Endocrinologia Basica y Clínicas 2011, 9na edicion; 493-500
- Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative con- formational disorder associated with AB, protein misfolding, and pro- teasome inhibition. Neurology 2006;66(suppl 1):S39–S48.
- Dalakas MC. The molecular and cellular pathology of inflammatory muscle diseases. Curr Opin Pharmacol 2001;1:300–306.
- Yunis EJ, Samaha FJ (1971) Inclusion body myositis. Lab Invest 25,240-248
- Askanas V, Engel WK. Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuro- pathol Exp Neurol 2001;60:1–14.
- Askanas V, Engel WK. New advances in inclusion-body myositis. Curr Opin Rheumatol 1993;5:732–741.
- Cupler J, Leon-Monzon M, Miller J, Semino-Mora C, Anderson TL, Dalakas MC. Inclusion body myositis in HIV-1 and HTLV-1 infected patients. Brain 1996;119:887–1893.
- Askanas V, Engel WK. Molecular pathology and pathogenesis of inclusion-body myositis. Microscopy Res Tech 2005;67:114–120.