Nota de los Editores: Los XTs de la colestasis son xantomas bien demarcados, de color anaranjado (tal como se aprecian en las imágenes), que a menudo aparecen en manos y pies. Se asocian principalmente con la CBP y la atresia biliar. En estas condiciones, el colesterol libre no puede ser excretado correctamente a la bilis y se regurgita hacia el plasma, donde se une a la albúmina y a los fosfolípidos para formar una lipoproteína anómala denominada lipoproteína X. Las personas afectadas poseen unos niveles de colesterol elevados (> 500mg/dl). Asimismo pueden presentar otros tipos de xantomas como los xantelasmas, los tuberoeruptivos y los tuberosos.
INFORMACIÓN BÁSICA: Colestasis crónica.-Cirrosis Biliar Secundaria.- Síndrome de Lipoproteína X.- (OMIM:-/ORPHA:-/CIE-10:-) .- El síndrome colestásico se caracteriza por una obstrucción de la vía biliar, ya sea intra o extra hepática, siendo la hipercolesterolemia una manifestación frecuente. El reflujo de la bilis ya sean intra o extrahepáticos, por el reflujo de colesterol biliar y/o de fosfolípidos o por alteración como es la deficiencia familiar de la enzima lecitina-colesterol aciltransferasa (LCAT). En los pacientes con colestasis causaría la gran acumulación de colesterol, que induce a su vez la formación de la denominada Lipoproteína X (LpX), esta es una vesícula bicapa compuesta que transporta este exceso de colesterol. La LpX es una LP anómala que comúnmente emerge y se acumula en circulación en pacientes con cirrosis biliar primaria, hígado obstructivo enfermedad y deficiencia de LCAT. La elevación severa de LpX en la circulación puede causar hipercolesterolemia que se asocia con xantomas cutáneos generalizados.
La lipoproteína X (LpX) es una fracción de lipoproteína anormal que puede detectarse en pacientes con hipercolesterolemia grave y enfermedad hepática colestática. LpX se compone principalmente de fosfolípidos y colesterol libre, con pequeñas cantidades de triglicéridos, ésteres de colesterilo y proteínas. No existen métodos ampliamente disponibles para la medición directa de LpX en la práctica de laboratorio de rutina.
La LpX tiene una estructura vesicular. Debido a la alta proporción de lípidos anfipáticos de la superficie (colesterol no esterificado y fosfolípidos) a los lípidos neutrales del núcleo, LpX forma una bicapa de fosfolípidos o incluso un fosfolípido multilaminar, disposición que da como resultado su apariencia similar a una cebolla en microscopía electrónica. Las partículas de LpX son de tamaño heterogéneo (30-100 nm) y pueden tener una densidad entre LDL y VLDL. La LpX es bastante heterogénea compuesta de fosfolípidos (lecitina) (66%), colesterol libre no esterificado (22%), albúmina y Apo C (6%), TGs 3%, y ésteres de colesterol (6%), y tiene un tamaño similar a las VLDL, aunque con la densidad de las LDL. La albúmina es un componente proteico importante y se encuentra principalmente dentro del núcleo acuoso de LpX. También contiene cantidades relativamente pequeñas de apolipoproteínas intercambiables , como apoA-I, apoE y apoC, presumiblemente unidas a su superficie de fosfolípidos, pero LpX no contiene apoB. A diferencia del LDL, que es un producto del catabolismo de la VLDL, la LpX es sintetizada directamente por los hepatocitos. LpX no contiene Apo B (el ligando primario para receptores de LDL hepáticos), quizás por eso el riesgo es relativamente bajo de ECV aterosclerótica. El aclaramiento de la LpX está mediado por el sistema reticuloendotelial esplénico no ligando-receptor, interfiriendo con la degradación de los QMs, y aumentando el riesgo de aterosclerosis.
El síndrome de LpX es un trastorno poco frecuente de hipercolesterolemia secundaria severa que se describió por primera vez en la década de 1950 y se relacionó con la enfermedad hepática colestásica. Se caracteriza por niveles elevados de CT y cLDL con TGs relativamente normales y, excepcionalmente, xantomas cutáneos, tromboembolismo por colesterol en la retina, depósitos corneales, pseudohiponatremia, neuropatía y síndrome de hiperviscosidad.
El síndrome de LpX aunque se identificó por primera vez en pacientes con obstrucción biliar extrahepática por enfermedad de la vesícula biliar, el síndrome de LpX se ha informado en el contexto de obstrucción biliar intrahepática (CBP y CEP), administración intravenosa de emulsión intralípida para nutrición parenteral total, trasplante de médula ósea, niños con disfunción hepática prematura y en la deficiencia hereditaria de LCAT.
Los informes del síndrome de LpX se describen en pacientes con colestasis. En estos pacientes, Manzato et al., postularon que LpX se forma como consecuencia del reflujo biliar al plasma, dando como resultado complejos de ácido biliar y albúmina creando un complejo de LPs similar en densidad al cLDL. Esto fue respaldado por experimentos in vitro en los que la bilis humana combinada con suero completo que contiene albúmina de voluntarios sanos dio como resultado la formación de LpX con un patrón de migración de electroforesis en gel similar a LpX en suero de pacientes con enfermedad hepática colestásica. La formación de LpX se volvió a demostrar en muestras venosas periféricas de sangre de perros tan pronto como tres horas después de crear anastomosis artificiales desde el conducto biliar hasta la vena cava inferior.
Paradójicamente, la síntesis de colesterol se intensifica en el contexto del síndrome de LpX. Se han sugerido varias teorías sobre este mecanismo, incluida la absorción del colesterol de los hepatocitos en partículas LpX y, por lo tanto, un aumento de la síntesis hepática de colesterol posiblemente mediada por la regulación inapropiada de 3-hidroxi-3-metil-glutaril-coenzima A (HMGCoA) reductasa. La comprensión actual de la formación de LpX a través de los complejos de albúmina y ácido biliares se cuestiona sobre la base de estas hipótesis y los datos recientes muestran cantidades insignificantes de ácido biliar en partículas LpX. Heimerl et al. sugieren que la relativa ausencia de inhibición por retroalimentación sobre la síntesis de colesterol de LpX regula positivamente el colesterol y la síntesis de fosfolípidos en hepatocitos que luego se liberan en la circulación como LpX. El mecanismo exacto aún no se ha determinado.
En varios casos de LpX debido a una colestasis por obstrucción biliar intra o extrahepática, se demuestra la resolución de los niveles de colesterol y cLDL después del tratamiento de la enfermedad subyacente o el trasplante de hígado. Sin embargo, aunque en exceso, el impacto clínico y la aterogénesis del colesterol elevado en forma de LpX siguen siendo poco claros.
Una revisión sistemática de tres ensayos prospectivos de pacientes con colangitis biliar primaria y síndrome de LpX señaló que, en un promedio de 10 años, el número y la causa de las muertes no fueron desproporcionadamente debido a causas cardíacas. Sin embargo, estos fueron todos estudios relativamente pequeños de pacientes con un solo tipo de enfermedad, lo que limita la generalización de este hallazgo.
La presencia de la Lp-X agrava la acumulación de colesterol plasmático por un lado, debido a que carece de la capacidad de inhibir la síntesis de colesterol, por falta de retroalimentación negativa, y por el otro, debido a que estimula la actividad de la HMG-CoA reductasa, enzima clave en su biosíntesis hasta cinco veces.
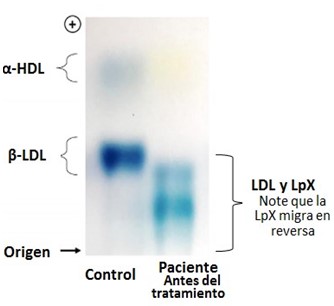
A nivel de laboratorio, la LpX se manifiesta en la electroforesis de Lps del plasma como una banda de movilidad cero a gamma, que presenta tinción metacromática con negro Sudán. Son estas partículas vesiculares las que hacen que el contenido sérico de fosfolípidos y colesterol no esterificado sea extremadamente alto. Otros datos incluyen, 1) ausencia de a-lipoproteína, lo que indica una baja concentración de HDL; 2) baja concentración de b-lipoproteína, lo que indica baja importante de la fracción LDL; 3) presencia de un banda por debajo de la b-lipoproteína, un patrón similar al reportado previamente para los casos de LpX (base la Figura 39).
Otras especies anormales de LPs, las denominadas Lp-Y, contiene cantidades apreciables de TGs y de apo B. Las LDL en la colestasis también contienen una cantidad extraordinariamente alta de TGs. Este cambio estructural supone unas LDL pequeñas y densas, aterogénicas.
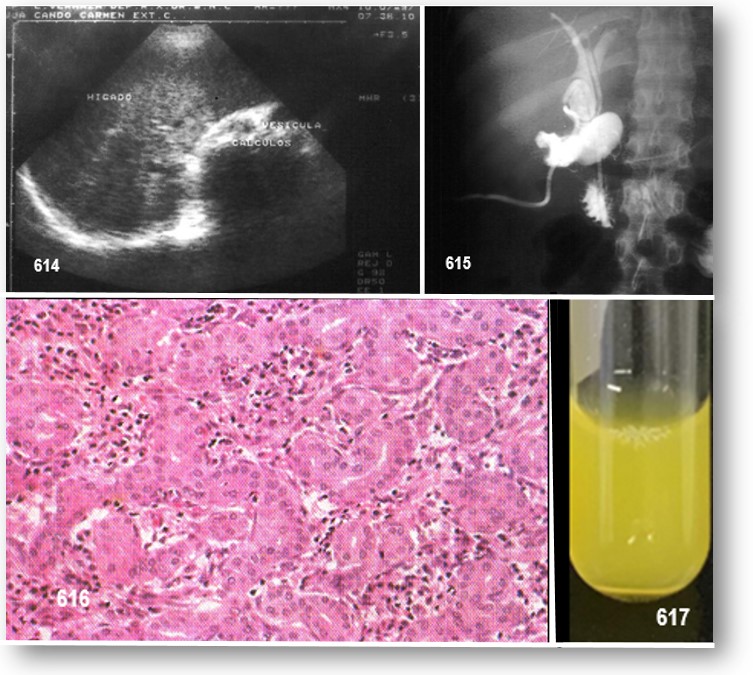
La hiperlipidemia asociada con la obstrucción del flujo biliar es compleja. La concentración de CT en estos pacientes exceden los 400mg/dl por lo general se asocian con obstrucción extrahepática o con tumor intrahepático. Hay varios tipos de LPs anormales. La más abundantes es la LpX.
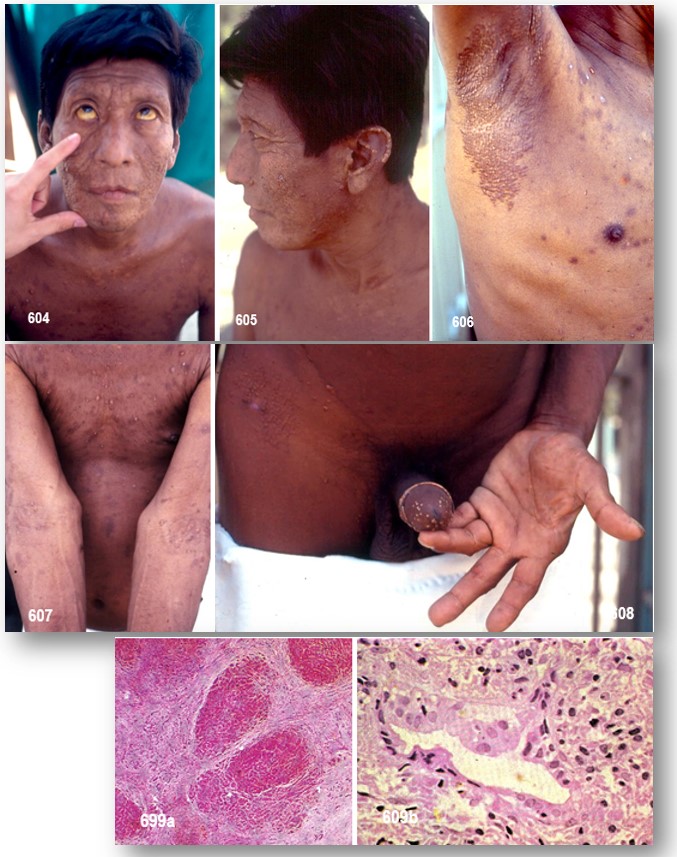
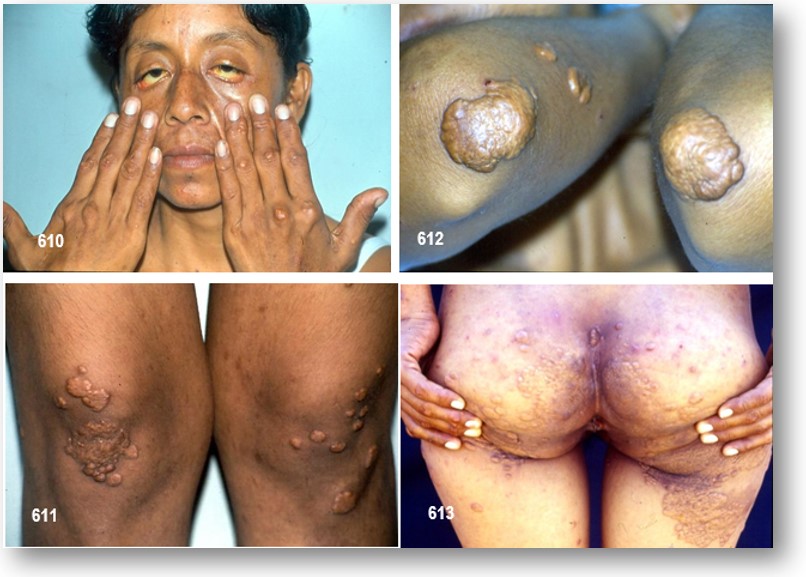
Los pacientes pueden tener XTs planos, especialmente en sitios de traumatismo leve, y XTs en los pliegues palmares. En ocasiones se encuentran XTs eruptivos. La afección xantomatosa de los nervios puede conducir a síntomas de neuropatía periférica, y las LPs anormales pueden ser aterogénicas. Mientras que la concentración de bilirrubina es casi normal en algunos pacientes con colestasis crónica, todos tienen actividad alta fosfatasa alcalina sérica. La neuropatía o la evidencia de aterosclerosis es la principal indicación para el tratamiento de la hiperlipidemia. Las resinas tienen cierto valor, mientras que los fibratos pueden ocasionar un incremento del CT. Las estatinas pueden lesionar el tejido hepático funcionante y, dado que su secreción es preponderantemente biliar, pueden producir miopatia al alcanzar concentraciones elevadas en la sangre. En casos resistentes la LDLaféresis es la alternativa. Estan indicadas vitamina liposolubles (A, D, E y K) en grandes dosis para superar el deterioro grave de la absorción.
Daño tisular generado por la LpX.- Niveles elevados de lípidos circulantes, incluidos LDL y lipoproteína rica en triglicéridos (VLDL), promueve la acumulación de lípidos intracelulares y en monocitos, lo que lleva a formación de monocitos espumosos. En los casos que presentamos, asumimos que la acumulación de LpX en la circulación inducide formación de monocitos, fenotipos modificados de monocitos y mejor adhesión de monocitos y absorción de LDLox.
A la luz de los hallazgos indican que los monocitos pueden ser un componente inmunológico sensible en pacientes con LpX, que contribuyen a la inflamación y el daño tisular.
La acumulación de lípidos intracelulares puede conducir a cambios fenotípicos en los monocitos. Un cambio es la regulación al alza de receptores de barrido (scavenger), y CD36, con captación mejorada de las LDL modificadas, la principal lipoproteína que provoca la formación de células espumosas en las lesiones ateroscleróticas. En consonancia con esto, la LpX excesiva genera el aumento de la formación de monocitos espumosos acompañado por una mayor expresión de receptores scavenger, que se correlacionó con aumento de la captación de LDLox.
Otros cambios fenotípicos en monocitos asociados con la acumulación de lípidos intracelulares incluye la regulación de moléculas de expresión y adhesión de citocinas como CD11c, una integrina b2 que juega un papel importante en adhesión de monocitos a células endoteliales y aterosclerosis. Sin embargo, en contra se a observado una regulación a la baja de varias citocinas proinflamatorias y CD11c en / sobre monocitos en el paciente con LpX antes del tratamiento en comparación con el participante de control. Se desconocen los mecanismos de estos cambios inesperados.
El grupo de Lian Zeqin especulan que los monocitos con grandes cantidades de lípidos acumulados y posiblemente el aumento de la inflamación y CD11c puede haber migrado a los tejidos rápidamente. Las alteraciones en los monocitos pueden haber contribuido a la inflamación de los tejidos con la formación de xantomas planos y eruptivos observado en ambos casos que publicamos.
El manejo de la dislipidemia a LpX en las formas agudas de colestasis el tratamiento de la dislipemia se basa en resolver la causa de base (véase caso CBS), por ejemplo, solucionar una obstrucción biliar. Por el contrario, en ciertas formas crónicas el manejo de las dislipidemias en sí presenta consideraciones especiales: en la CBP, la colestiramina o el ácido ursodeoxicólico (Ursofalk®/caps: 250mg) mejora el prurito, la colestasis y disminuye el colesterol plasmático en un 16-19%, sin ningún cambio en los niveles de cHDL o TGs. Alternativamente fibratos asociado a ácido ursodeoxicólico. Sin embargo, la terapia farmacológica de la hipercolesterolemia por LpX difiere de la de la hipercolesterolemia poligénica o monogénica convencional, que se basa principalmente en estatinas, ezetimiba e inhibidores de PCSK9. La razón es que ninguno de estos medicamentos se dirige a la fisiopatología subyacente y, por lo tanto, carece de eficacia en este contexto.
En el caso de las estatinas, la supresión de la síntesis de colesterol en el hígado induce una regulación positiva del RLDL, pero como LpX no tiene apoB, las estatinas no afectarán la eliminación de esta lipoproteína por el hígado. Además, como la mayoría de las estatinas se eliminan a través de la bilis, los pacientes con enfermedades colestásicas basales pueden alcanzar concentraciones de estatinas tóxicas.
En el caso de la ezetimiba, este agente previene la absorción intestinal del colesterol de la dieta, que tiene una participación marginal en la conformación de LpX. Además, los pacientes con colestasis ya tienen una absorción muy limitada de colesterol en la dieta debido a la formación insuficiente de micelas, típica de estas enfermedades.
Se puede considerar el uso de fibratos en pacientes con hipercolesterolemia LpX. Aunque los fibratos son esencialmente una terapia para la hipertrigliceridemia, tienen efectos anti-colestáticos, antiinflamatorios y antifibróticos en enfermedades hepáticas, especialmente en pacientes con CBP. Los estudios de monoterapia con fibratos o en combinación con ácido ursodesoxicólico han demostrado mejoras significativas en los marcadores de daño hepatocelular. No obstante, el uso de fibratos todavía no se considera una terapia de primera línea para LpX.
Esta hepatopatía no representa contraindicación absoluta pero si de cuidados en la prescripción de estatinas o fibratos, ya que ambos fármacos han demostrado tanto una mejoría del perfil lipídico como también del hepatograma.
La reducción farmacológica de LpX con estatinas tiene poca utilidad, pero se ha intentado con cierto éxito. Teniendo en cuenta el aumento paradójico de la síntesis de colesterol, las estatinas podrían reducir potencialmente la formación de LpX, pero su mecanismo principal para reducir el cLDL (regulación vía de RLDL) puede no ser tan importante en esta condición ya que estas partículas (LpX) no parecen eliminarse por RLDL. Es de destacar que varias estatinas, lovastatina (Lipivas®), simvastatina (Zocor®), atorvastatina (Lipitor®) y rosuvastatina (Crestor®) se eliminan en la bilis y pueden acumularse en dosis tóxicas en casos de colestasis.
Cuando un paciente con LpX elevada presenta complicaciones graves como síndrome de hiperviscosidad, embolia pulmonar o colesteroloma, la plasmaféresis es la terapia complementaria preferida. Sin embargo, debe tenerse en cuenta que la aféresis de LDL no se recomienda para la eliminación de LpX, ya que la ausencia de apoB en LpX hace que esta terapia sea ineficaz. La plasmaféresis se ha empleado no solo en pacientes con enfermedad hepática primaria, sino también en pacientes con enfermedad de injerto contra huésped hepático. Sin embargo, su uso debe considerarse sólo como una medida temporal, como la única terapia definitiva en el caso de enfermedades hepáticas primarias es trasplante de hígado. Contrario a esto hay tres informes de casos de plasmaféresis con resultado exitoso de los niveles de LpX y la normalización de los paneles lipídicos en pacientes con colangitis esclerosante primaria y uno con síndrome de LpX inducido por enfermedad injerto contra huésped. Debido a que esta es una condición rara, el tratamiento con plasmaféresis se lo reserva para el desarrollo de síntomas más severos (p. Ej., Síndrome de hiperviscosidad), tal como lo mencionamos anteriormente.
Bibliografía
- Gower RM, Wu H, Foster GA, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31:160–166.
- Tama Viteri FA. Clínica y Terapéutica de las Dislipidemias. Primera Edición. Universidad de Guayaquil (UG) Eduqil, 2011.
- Lian Z, Perrard XD, Peng X, et al. Replacing saturated fat with unsaturated fat in western diet reduces foamy monocytes and atherosclerosis in male Ldlr(-/-) mice. Arterioscler Thromb Vasc Biol. 2020;40:72–85.
- https://www.researchgate.net/publication/351708586_SINDROME_DE_LIPOPROTEINA_X
- Wu H, Gower RM, Wang H, et al. Functional role of CD11c1 monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717.
- Bernelot Moens SJ, Neele AE, Kroon J, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. 2017;38:1584–1593.
- Khan IM, Pokharel Y, Dadu RT, et al. Postprandial monocyte Activation in individuals with metabolic syndrome. J Clin Endocrinol Metab. 2016;101:4195–4204.
- Kashyap SR, Ioachimescu AG, Gornik HL, et al. Lipid-induced insulin resistance is associated with increased monocyte expression of scavenger receptor CD36 and internalization of oxidized LDL. Obesity (Silver Spring). 2009;17:2142–2148.
- Yla-Herttuala S, Palinski W, Rosenfeld ME, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–1095.
- Manzato E, Fellin R, Baggio G, Walch S, Neubeck W, Seidel D. Formation of lipoprotein-X. Its relationship to bile compounds. J Clin Invest 1976; 57: 1248-60.
- Heimerl S, Boettcher A, Kaul H, Liebisch G. Perfil lipídico de la lipoproteína X: implicaciones para la dislipidemia en la colestasis . Biochim Biophys Acta . 2016; 1861 : 681-687.
- Phatlhane DV, Zemlin AE. Hipercolesterolemia severa mediada por lipoproteína X en un paciente con colestasis . Ann Hepatol . 2015; 14 : 924-928.
- Cuperus FJC, Halilbasic E, Trauner M. Tratamiento con fibratos para la cirrosis biliar primaria . Curr Opin Gastroenterol . 2014; 30 : 279-286.
- Wong ML, Raghavan RP, Hedger NA, Ellis RD, Meeking DR, Albon L. El uso de plasmaféresis en el manejo de la cirrosis biliar primaria que se presenta con hipercolesterolemia profunda . Br J Diabet Vasc Dis . 2012; 12 : 156-158
- Joukhadar R, Chiu K. Hipercolesterolemia severa en pacientes con enfermedad de injerto contra huésped que afecta al hígado después del trasplante de células madre . Endocr Pract . 2012; 18 : 90-97.
- Turchin A, Wiebe DA, Seely EW, Graham T, Longo W, Soiffer R. Hipercolesterolemia severa mediada por lipoproteína X en pacientes con enfermedad crónica de injerto contra huésped del hígado . Trasplante de médula ósea . 2005; 35 : 85-89.
- Cohen LB, Ambinder EP, Wolke AM, Field SP, Schaffner F. Papel de la plasmaféresis en la cirrosis biliar primaria . Gut . 1985; 26 : 291-294.