CASO 1
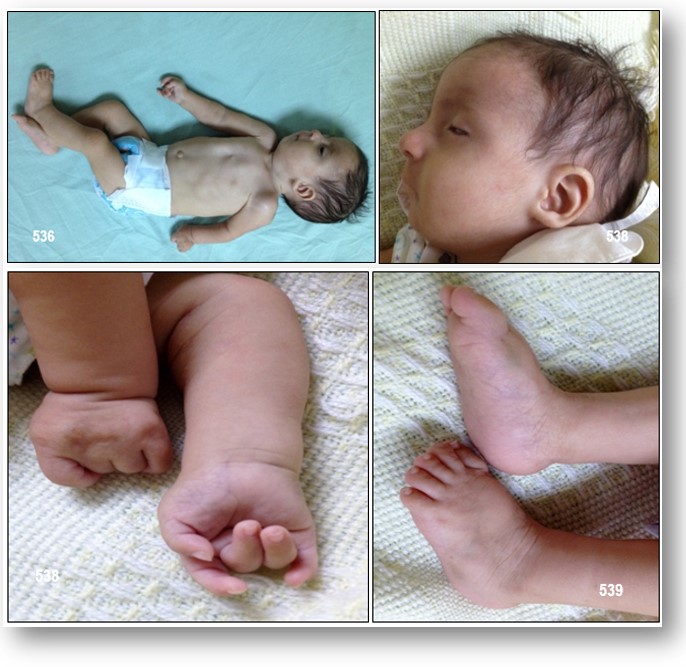
Nota de los investigadores: Se realizaron otras valoraciones complementarias: EEG: Hipsarritmia. Ecodoppler del corazón: Comunicación interventricular, persistencia del conducto arteriovenoso e hipertensión pulmonar moderada. En su hospitalización, la paciente permaneció 13 días, siendo manejada por el servicio de neurología con fármacos anticonvulsivantes; por el servicio de cardiología, con diuréticos. En su evolución presento cuadro respiratorio muy sugestivo de infección recibiendo antibióticos y terapia respiratoria de soporte, evolucionando favorablemente (Gentileza de la Dra. Verónica Del Roció Rosero Armijos. Médico Residente del Hospital de Niños Dr. Francisco De Icaza Bustamante. Guayaquil-Ecuador).
CASO 2
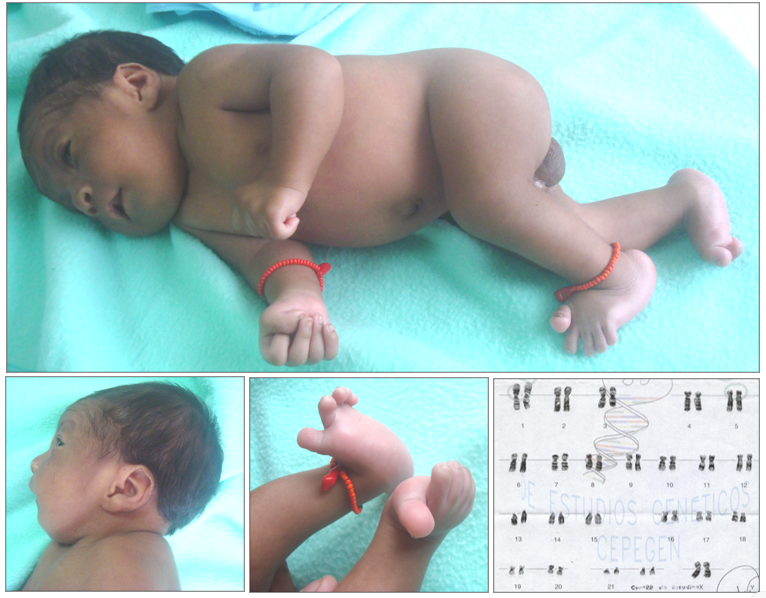
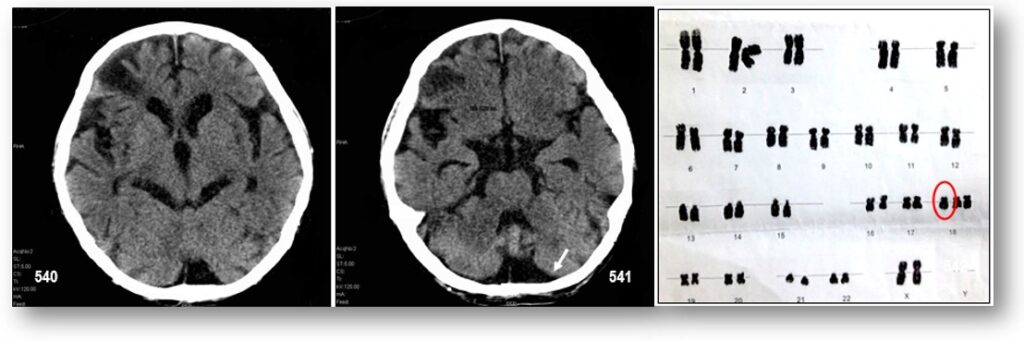
Imágenes secuenciales: Imagen 1. Sindrome de Edwards o Trisomía 18. – Paciente de 1 mes de nacido que presenta rasgos dismórficos: microcefalia, fontanelas amplias, occipucio prominente con diámetro bifrontal estrecho. Manos trisómicas (posición de las manos característica con tendencia a puños cerrados, con dificultad para abrirlos, y con el segundo dedo montado sobre el tercero y el quinto sobre el cuarto muy característico de la trisomía 18), uñas de manos y pies hipoplásicas. Talones prominentes con primer dedo de los pies corto y en dorsiflexión. Imagen inferior: El Cariotipo del paciente nos desconcertó, sin embargo, no descartamos la posibilidad que se trate de un mosaicismo en el cromosoma 18. Informe citogenético: Cariotipo Masculino normal: 46, XY.
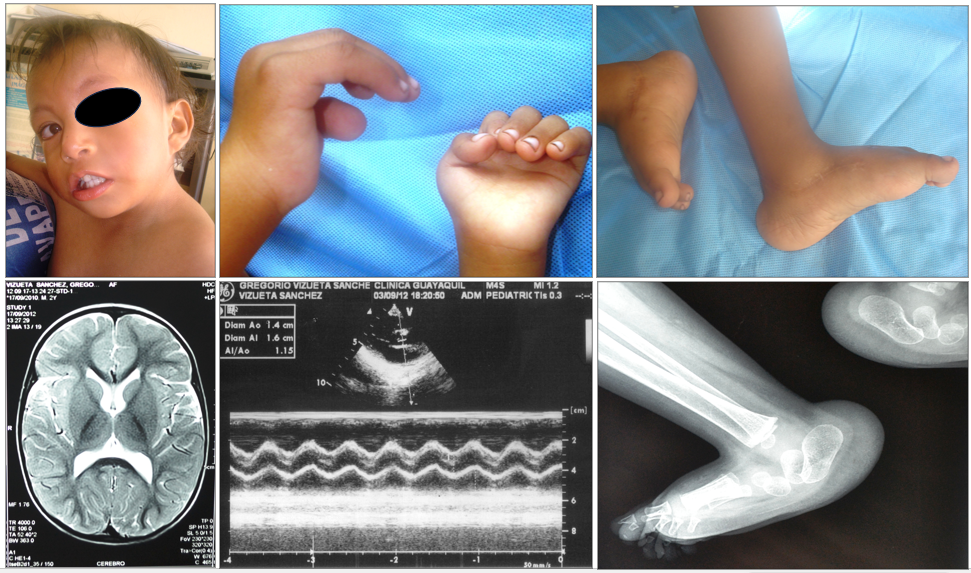
Imágenes secuenciales: Sindrome de Edwards o Trisomía 18. – Paciente de la imagen 1 dos años más tarde. Nótese que se mantienen las anomalías más relevantes del síndrome: cabello escaso, boca pequeña (en “carpa”), orejas de implantación baja, y a nivel de las manos, la mano trisómica (cabalgamiento de los ortejos) así como los pies zambo y calcáneo prominente; es de anotar que el paciente no ha desarrollado el habla y mantiene estado hipotónico y retraso mental. Ecodoppler de.l corazón: Trazado de Ecodoppler Modo M y Bidimensional: No cardiopatía estructural. Trazado Normal. RMN de cerebro simple y contrastado: cortes potenciales axiales en t1, t2, flair axiales y t1 sagitales, coronales normales. Radiografías simples de: Tórax. Abdomen. Miembros inferiores: Pies zambos con talones prominentes por crecimiento anormal del calcáneo. Screening ecosonografiico del abdomen.
INFORMACIÓN BÁSICA.- TRISOMIAS: Síndrome de Edwards o Trisomia 18 (OMIM/ ORPHA:3380).- Síndrome polimalformativo, consecuencia de un imbalance cromosómico debido a la existencia de tres cromosomas 18. Se da en todas las razas y zonas geográficas. La mayoría de casos están asociados con trisomía 18 libre. Se ha detectado trisomía 18 en mosaico en unos pocos pacientes que presentaban un cuadro clínico que varía desde la trisomía 18 clásica hasta un fenotipo normal dependiendo del número de células trisómicas presentes en los tejidos. El fenotipo de la trisomía 18 parece estar asociado con la presencia de tres copias del intervalo 18q11-q12. Debido a su alta tasa de mortalidad en los recién nacidos, mas del 30% de los casos, se le considera una enfermedad de tipo letal.
Incidencia.- Su incidencia se estima entre 1/6.000 y 1/8.000 nacimientos vivos, aunque su incidencia real depende del porcentaje de diagnóstico prenatal mediante la realización o no de amniocentesis, siendo mucho más frecuente en abortos espontáneos y mortinatos. En más de un 95% de fetos con esta anomalía cromosómica se produce una muerte in útero. Por razones que se desconocen, el índice de supervivencia es mayor en mujeres que en hombres, lo que supone una predominancia femenina entre recién nacidos vivos con trisomía 18.
Etiología: En ocasiones, ocurre un error durante la formación del óvulo o del espermatozoide, y esto causa la presencia de un cromosoma 18 adicional. Cuando esta célula aporta el cromosoma 18 adicional al embrión, el resultado es la trisomía 18. El cromosoma 18 adicional puede provenir tanto del óvulo de la madre como del espermatozoide del padre. Las características de la trisomía 18 es el resultado de la presencia de este cromosoma 18 adicional en cada célula del cuerpo. El 95-96% de casos corresponden a trisomía completa producto de no-disyunción, siendo el resto trisomía por traslocación. La trisomía parcial y el mosaicismo para trisomía 18 suelen presentar un fenotipo incompleto, con ausencia de algunas de las anomalías típicas del Síndrome de Edwards. El fenotipo de la trisomía 18, como la trisomía 21, puede resultar de varios cariotipos raros distintos de la trisomía completa. Puede existir una traslocación que involucre todo el cromosoma 18 o la mayor parte de el, que puede ser de novo o heredada de un progenitor portador equilibrado. La trisomía también puede encontrarse también en forma de mosaico con expresión variable, pero generalmente algo más leve.
No se ha identificado una región crítica cromosómica única, responsable del síndrome. Parece que es necesaria la duplicación de dos zonas, 18q12-21 y 18q23 para que se produzca el fenotipo típico de Síndrome de Edwards, con una zona, 18q12.3-q21.1 con fuerte influencia en el retraso mental.
Tipos de Trisomía 18
- – Trisomía 18 Completa. Es el primer tipo, y se llama “Trisomía 18 Clásica o Completa”. Significa que existen 3 copias completas del cromosoma 18 en cada una de las células del paciente. Si todas las células (5 ó 20 células dependiendo del protocolo del laboratorio) que examinaron tienen el cromosoma 18 extra y está completo, se dice que es una trisomía completa.
- – Trisomía 18 en Mosaico (mosaicismo). Este tipo de trisomía se refiere a un paciente que tiene algunas células con 3 copias del cromosoma 18 y algunas células con las 2 copias normales de ese cromosoma. El efecto del mosaicismo es amplio y variado. Los individuos afectados pueden tener todos los problemas de una Trisomía Completa o ninguno y cualquier cosa intermedia, aunque por lo general las características clínicas comunes de la trisomía 18 pueden ser más leves cuando se presenta en mosaico, puesto que no todas las células del cuerpo son portadoras de un cromosoma extra. Alrededor de 10% de las personas con trisomía 18 la tienen en mosaicismo. Sin embargo, el riesgo de muerte prematura es exactamente igual ya que no es la anomalía cromosómica en sí la que causa la muerte sino la forma en que se afecta al bebe en desarrollo por los cambios que hay en el material genético de sus células. El efecto verdadero del mosaico no depende del número de células afectadas sino de su distribución. El mosaicismo, aunque aún no se sabe con certeza como ocurre, se tienen las siguientes teorías:
- – Teoría número 1. La más popular es la que considera que tanto el óvulo como el espermatozoide contenían el complemento “normal” de cromosomas así que la “meiosis” fue normal. Sin embargo, poco después de que se realizó esta división, esto es, después de que el óvulo fue fertilizado por el espermatozoide y empieza a dividirse para formar el embrión, hay un error en esta nueva división y terminarán algunas células con un cromosoma extra y otras serán normales. La distribución de las células con la anomalía cromosómica dependerá de cuándo exactamente ocurrió el error original.
Si ocurrió antes de la segunda semana de gestación – que es cuando el huevo fertilizado empieza a diferenciar los sistemas del cuerpo- entonces la distribución será más pareja en todos los sistemas del organismo, si ocurre después, entonces la distribución será más dispareja y algunos sistemas tendrán un porcentaje mayor de células afectadas que otros. De hecho, algunos sistemas podrán estar totalmente “sanos”.
– Teoría número 2. Esta es menos popular y se le llama “rescate celular”. En esta teoría se considera que el bebé empezó su vida con todas sus células afectadas por la Trisomía pero como el cuerpo está constantemente tratando de corregir cualquier error que se presente, en un momento dado fue capaz de hacerlo y se descartó el cromosoma extra. La división de las células continuó como de “células con un número normal de cromosomas”. Sin embargo, se considera que hubo algunas células que no fueron capaces de corregir ese error y continuaron así su división. En este caso también, la distribución es variable y depende de cuándo y en dónde se realizó la corrección.
El mosaicismo se puede diagnosticar de diferentes maneras. A veces, las deferentes células en la sangre tienen diferente composición cromosómica. Para esos individuos, un análisis de sangre puede detector el mosaicismo. En otros, el diagnóstico es más difícil. Por ejemplo, un análisis de sangre puede diagnosticar mosaicismo si el cambio cromosómico está ubicado en las células de la piel. En casos como este, es necesaria alguna prueba adicional para detectar mosaicismo. Por lo general, puede incluir el examen de otros tejidos del cuerpo, como la piel. Un genetista puede ayudar a determinar la necesidad de hacerlo.
– Trisomía 18 parcial. En algunos casos puede haber una copia extra de una porción del cromosoma 18. Los cromosomas en realidad están formados por dos partes llamadas “brazos” unidas por una parte más estrecha o centro que es muy importante en la división celular.
Un brazo es más corto (llamado P por pequeño) que el otro (llamado Q porque es la letra que sigue en el abecedario) y contienen a los genes o responsables de la herencia correspondientes a este cromosoma.
En realidad existen diferentes tipos de trisomía parcial 18. Algunas personas tienen una duplicación del brazo corto del cromosoma.
Otras personas pueden tener una duplicación del brazo largo del mismo. O, una persona puede tener una duplicación de parte del brazo largo y de parte del brazo corto del cromosoma. Por consiguiente, dos personas con “trisomía parcial 18” pueden tener dos trastornos diferentes, dependiendo de qué parte del cromosoma esté involucrada.
La parte del cromosoma que está duplicada generalmente no está sola sino que se ha unido a otro cromosoma, por ejemplo, puede estar unido al cromosoma 9.
Traslocación: A menudo, pero no siempre, las Trisomías Parciales reflejan una Traslocación. Existen dos tipos de Traslocaciones: recíproca y Robertsoniana.
Los cromosomas son muy frágiles y pegajosos. En algunas ocasiones, durante la mitosis los cromosomas se rompen y a veces se vuelven a unir sin problemas, pero a veces no se unen en la parte que se rompieron. A esto se le llama una “Traslocación Balanceada”. En circunstancias normales esto no causa problemas ya que todo el material genético se encuentra ahí pero en diferente orden. A las personas que tienen una traslocación se les llama “portadores” y generalmente nunca se dan cuenta de esta situación sino hasta que ellos o alguno de sus hermanos tienen hijos.
Cuando una persona con Traslocación tiene hijos, es posible que el óvulo o el esperma tengan una distribución desbalanceada del material genético y por lo tanto se incrementan los riesgos de tener un bebé con anomalías cromosómicas.
La Traslocación Robertsoniana es un tipo diferente de traslocación y afecta únicamente a los cromosomas 13, 14, 15 y 21 y es bastante común. Sin embargo cuando estas personas producen un óvulo o un espermatozoide, tendrán una ganancia o una pérdida de material genético lo que resultará en una trisomía completa.
Manifestaciones Clínicas: En las primeras semanas de vida se presentan hipotonía, hiporreactividad y problemas de alimentación (mala succión), seguidos por una progresión a hipertonía, con niños que presentan una falta aparente de conciencia del entorno. Entre las características comunes se encuentran retraso del crecimiento intrauterino y postnatal, aspecto demacrado con hipotrofia, microcefalia con cráneo estrecho y dolicocefalia, microrretrognatia, hipertelorismo y orejas angulares y anormalmente modeladas.
- – Retraso de crecimiento pre y postnatal: (peso medio al nacer: 2.340 g). La Tabla1 resume toda la clínica del síndrome.
- – Nacimiento postérmino
- – Retraso mental con falta de desarrollo psico motor: Importante retraso mental con incapacidad para que el bebé adquiera capacidades mentales, verbales y motoras.
- – Panículo adiposo y masa muscular escasa al nacer.
- – Hipotonía inicial que evoluciona a hipertonía.
- –Craneofaciales. Características faciales: Esta condición del cerebro puede ser causa de los cambios que se observan en la cara del bebé en donde los ojos están demasiado separados (hipertelorismo ocular), ptosis de los párpados superiores y puede haber estrabismo y una respuesta disminuida a los estímulos de luz; la nariz no está bien formada y parece respingada. Las orejas están implantadas muy abajo y tienen una forma inusual. La boca es pequeña con un el paladar muy estrecho y micrognatia. El labio y paladar hendido se encuentran en un 60 a 80% de los bebés con este síndrome.
Características Craneales: microcefalia, fontanelas amplias, occipucio prominente con diámetro bifrontal estrecho, defectos oculares (opacidad corneal, catarata, microftalmía, coloboma de iris), fisuras palpebrales cortas, orejas displásicas de implantación baja, micrognatia, boca pequeña, paladar ojival, labio/paladar hendido.
– Extremidad superior: mano trisómica (posición de las manos característica con tendencia a puños cerrados, con dificultad para abrirlos, y con el 2do dedo montado sobre el 3ro y el 5to sobre el 4to), uñas de manos y pies hipoplásicas. hipoplasia/aplasia radial.
–Extremidades inferiores.- Entre las anomalías de los pies se incluyen pies zambos y/o pies en mecedora (piernas cruzadas, posición preferida), limitación a la extensión (>45º) de las caderas, talón prominente con primer dedo del pie corto y en dorsiflexión, sindactilia 2º-3 tercer dedos del pie. Puede haber sindactilia o polidactilia.
– Tórax-Abdomen: mamilas hipoplásicas, hernia umbilical y/ó inguinal, espacio intermamilar aumentado, onfalocele
– Urogenital: criptorquidia, hipoplasia labios mayores con clítoris prominente, malformaciones uterinas, hipospadias, escroto bífido.
– Malformaciones renourológicas: riñón en herradura, ectopia renal, hidronefrosis, duplicidad ureteral, riñón poliquístico.
– Cardiovascular: cardiopatía congénita* presente en 90% de casos (comunicación interventricular con afectación valvular múltiple, conducto arterioso persistente, estenosis pulmonar, coartación de aorta, transposición de grandes arterias, tetralogía de Fallot, arteria coronaria anómala; dextrocardia).
– Tracto gastrointestinal: divertículo de Meckel, páncreas ectópico, fijación incompleta del colon, ano anterior, atresia anal y/o esofágica.
– Sistema Nervioso Central: hipoplasia/Aplasia de cuerpo calloso, agenesia de septum pellucidum, circunvoluciones cerebrales anómalas, hidrocefalia, espina bífida (6% de los casos), así como epilepsias. Es común que la parte frontal del cerebro no se divida apropiadamente lo que resulta en una condición llamada “holoprosencefalia”. Al ser una entidad heterogénea, deben investigarse anomalías neuromusculares.
– Piel: cutis marmorata, hirsutismo en espalda y frente.
– Signos radiológicos: esternón corto con núcleos de osificación reducidos, pelvis pequeñas, caderas luxadas. Anomalías más frecuentes, que han demostrado su utilidad en el diagnóstico clínico y que se considera que están presentes en >50% de casos.
Otras anomalías: Arteria umbilical única. Pseudoquiste del cordón umbilical. Con menor frecuencia se observan labio/paladar hendido, artrogriposis, aplasia radial, espina bífida y anencefalia, holoprosencefalia y onfalocele.
Diagnóstico: Estudio citogenético (cariotipo que puede mostrar: trisomía 18, trisomía parcial o translocación). Las anomalías cromosómicas, como la trisomía 18, pueden diagnosticarse antes del nacimiento a través del análisis de las células del líquido amniótico, de la placenta o del cordón umbilical (biopsias), además a través de hallazgos ecográficos (retraso en el crecimiento, malformaciones, múltiples quistes del plexo coroideo, etc.) y puede confirmarse mediante análisis del cariotipo del feto.
Los marcadores séricos maternos (utilizados para el diagnóstico de la trisomía 21) también pueden ser anormales. Un diagnóstico rápido por hibridación de fluorescencia de interfase in situ (hibridación de fluorescencia in situ-FISH-) en muchos recién nacidos con trisomía 18 (y trisomía 13) puede ser de mucha ayuda para la toma de decisiones respecto a los cuidados e intervención quirúrgica. El análisis de linfocitos normalmente toma más de 48 horas, pero el análisis de una muestra de 24 horas puede ser importante en algunos casos donde se evalúan las decisiones respecto a cirugía. Más recientemente se han propuesto frotis bucales para el diagnóstico rápido utilizando sondas de ácido desoxiribonucleico (DNA).
Debido a la asociación entre trisomía 18 y edad materna avanzada, el diagnóstico prenatal de fetos con trisomía 18 ha ido en paralelo con el desarrollo de la amniocentesis y el programa de muestreo de vellosidades. En años recientes, se ha desarrollado una cantidad significativa de literatura alrededor de la detección sonográfica de patrones sugestivos de trisomía 18 (y trisomía 13).
Debido a la ocurrencia común de retardo en el crecimiento intrauterino en fetos con trisomía 18 en el segundo y tercer trimestre, el diagnóstico es hecho a menudo durante estos períodos gestacionales. Además, en un 30-60% de los embarazos con trisomía 18 ocurre polihidramnios y a menudo conduce a diagnóstico prenatal.
Se han descrito varios signos prenatales característicos en fetos con trisomía 18. Estos descubrimientos, en conjunto con retardo de crecimiento intrauterino o una malformación mayor consistente con el fenotipo, a menudo trae a colación el diagnóstico en ambientes prenatales después de 20 semanas de gestación. Hill (1996) revisó cuidadosamente la detección sonográfica de la trisomía 18. Los marcadores fonográficos más publicitados incluyen:
- – Quistes del plexo coroide,
- – Grandes cisterna magna y un
- – Calvarium de forma de frutilla.
Más recientemente, De Vorc (2000) documentó que 29 de 30 fetos de Segundo trimestre con trisomía 18 tuvieron “anatomía fetal anormal” con uno de estos marcadores o defectos del corazón. Tongsong et al. (2002) revisaron los rasgos fonográficos de mitad del embarazo de 25 casos y encontraron que todos tuvieron a lo menos un hallazgo, con restricción en el crecimiento de casi la mitad de ellos.
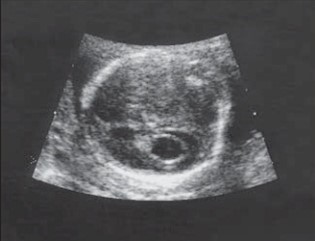
La utilidad de los quistes del plexo coroide como un signo de trisomía 18 es un tópico de controversia en la literatura obstétrica, sin un claro consenso sobre cuando ofrecer amniocentesis para los cariotipos cuando se descubren los quistes, particularmente cuando no hay otros descubrimientos Gross et al., 1995; Shields et al., 1996; Reinsch, 1997).
Debido a que algunos fetos de segundo trimestre con trisomía 18 no tienen otras anomalías o descubrimientos de crecimiento, hay un riesgo creciente de trisomía 18 en la presencia de un quiste aislado del plexo coroide (Reinsch. 1997).
Sin embargo, la decisión respecto al desempeño del diagnóstico invasivo prenatal es complicado, y el autor recomienda revisar los números de riesgo con la familia a la luz de los riesgos específicos de la edad materna y otros sondeos prenatales, Gratton et al. (1996) presentaron estimaciones basadas en la edad maternal y sondeos de marcadores múltiples en presencia de un quiste aislado del plexo coroide en el escenario clínico.
Más recientemente, Demasio et al. (2002) llevaron a cabo un meta-análisis de varias series de casos e indicaron ningún aumento del riesgo de trisomía 18 en mujeres bajo 35 años de edad. En el análisis final, es la familia la que tomará la decisión basada en el riesgo individual en el contexto del riesgo del procedimiento, el impacto percibido de trisomía 18, y el beneficio percibido de hacer el diagnóstico in útero.

Diagnóstico diferencial: Clínicamente puede plantearse con:
– Trisomía 13: Holoprosencefalia con fisura labial central, fisura palatina, frente en declive, microftalmia, coloboma de iris, hipotelorismo, puente nasal prominente, ulceraciones tipo aplasia cutis en cuero cabelludo, cuello corto, uñas estrechas e hiperconvexas, polidactilia en manos y pies, mamilas hipoplásicas, pies «en mecedora», talón prominente, hemangiomas capilares, dextrocardia. Cariotipo: trisomía 13.
– Secuencia de akinesia/hipokinesia fetal o Síndrome de Pena-Shokeir, ó Síndrome de Moessinger: Polihidramnios, movilidad fetal escasa, cordón umbilical corto, anomalías faciales como hipertelorismo, fisura palatina con paladar hendido, cuello corto, orejas displásicas, además artrogrifosis; camptodactilia, pies zambos, hipoplasia pulmonar, testes no descendidos, pliegues de flexión palmo-plantares hipoplásicos, escasa movilidad postnatal. Cariotipo: normal.
Evolución: Mortalidad del 95% en el primer año de vida. El 5% restante suele sobrevivir más tiempo (La tasa de mortalidad en los supervivientes es del 2% a los 5 años de vida, y un 1% de posibilidad de llegar a los 10 años de edad). Las niñas presentan mayor tasa de supervivencia. Causa principal de fallecimiento: cardiopatía congénita, apneas, y neumonía. Problemas más frecuentes en los supervivientes:
- – Dificultades en la alimentación: La mayoría necesitarán alimentación por sonda. Puede ser necesario recurrir a la gastrostomía. No obstante algunos consiguen tomar bien el biberón, y se ha conseguido lactancia materna en casos aislados. Muy pocos serán capaces de comer solos.
- – Escoliosis: Puede afectar mucho la calidad de vida de los supervivientes. No parecen ser de utilidad los aparatos ortopédicos, lo mejor y más cómodo es usar almohadones o respaldos de madera que se coloca en la cuna ó en el carrito para modificar la postura del niño
- – Estreñimiento: Precisarán enemas
- – Infecciones: Neumonía, otitis media, e infecciones urinarias.
- – Desarrollo psíquico/motor: Importante retraso. En un grupo de supervivientes con edad media cronológica de 8 años, la edad de desarrollo media fue de 6.8 meses.
Pueden llegar a ser capaces de utilizar 4 ó 5 palabras
Tratamiento: El tratamiento sólo es de apoyo. El tratamiento quirúrgico de las malformaciones apenas mejora el pronóstico desfavorable asociado con este síndrome: el 90% de niños muere en el primer año de vida por complicaciones cardiacas, renales o neurológicas, o por repetidas infecciones. Se ha registrado supervivencia prolongada (en algunos casos hasta edad adulta), principalmente en casos de trisomía en mosaico o parcial (resultante de la translocación). La mayoría de pacientes sin mosaicismo sólo desarrollan autonomía limitada (ausencia de habla y deambulación). El retraso en el crecimiento es significativo.
Consejo genético: La trisomía 18 suele darse de forma aislada en familias por otra parte normales.
En estos casos el riesgo de recurrencia estimado es del 0.55%. En los casos de trisomía por traslocación, los padres deben ser remitidos a un Servicio de Genética para estudio citogenético. Es más frecuente en madres de edad avanzada. A partir de los 35 años la frecuencia aumenta progresivamente desde 1/2500 nacidos vivos a los 36 años hasta 1/500 a los 43.
En mujeres de edad >35 años, ó con hijo anterior con trisomía 18 debe ofrecerse diagnóstico prenatal mediante amniocentesis en los siguientes embarazos. Existe gran variabilidad en el desarrollo físico y psíquico en los supervivientes a medio-largo plazo. Los pocos pacientes que alcanzan largas supervivencias tienen marcadas limitaciones psicomotoras.
En otros, el diagnóstico es más difícil. Por ejemplo, un análisis de sangre puede diagnosticar mosaicismo si el cambio cromosómico está ubicado en las células de la piel. En casos como este, es necesaria alguna prueba adicional para detectar mosaicismo. Por lo general, puede incluir el examen de otros tejidos del cuerpo, como la piel. Un genetista puede ayudar a determinar la necesidad de hacerlo.
Bibliografía
- Demasio K, Canterino J, Ananth C, Fernandez C, Smulian J, Vintzileos A. Isolated choroid plexus cysts in low-risk women less than 35 years old. Am J Obstet Gynecol. 2002;187:1246–1249.
- Edwards, JH, Hamden DG, Cameron AH, Crosse VM, Wolff OH. A new trisomic syndrome. Lancet. 1960;1(7128):787-90.
- Gratton RJ, Hogge WA, Aston CE (1996). Choroid plexus cysts and trisomy 18: Risk modification based on maternal age and multiple-marker screening. Am J Obste Gynecol 175: 1493-1497
- DeVore GR. (2000) Second trimester ultrasonography may identify 77 to 97% of fetuses with trisomy 18. J Ultrasound Med. 19:565-576.
- Gross SJ, Shulman LP. Tolley EA, Emerson DS, Felkcr RE, Simpson JL, Elias S (1995) Isolated fetal choroid plexus cysts and trisomy 18: A review and meta-analysis. Am J Obstel Gym-Col 172:83-87.
- Hill LM. La detección ecográfica de las trisomías 13, 18 y 21. Clin Obstet Gynecol. 1996; 39 : 831 – 850. Doi: 10.1097 / 00003081-199612000-00011.
- Shields LE, Uhrich SB, Easterling TR, Cyr DR, Mack LA (1996) Isolated fetal choroid plexus cysts and karyotype analysis: Is it necessary-./ Ultrasound Med 15:389-394.
- T Tongsong et al. Sonographic features of trisomy 18 at midpregnancy Journal of Obstetrics and Gynecology Research. Volumen 28, Número 5, páginas 245-250, octubre de 2002
- Reinsch RC (1997) Choroid plexus cysts: an association with trisomy. Am J Obstet Gynecol 176:1381-183.
- Gardner RJM, Sutherland GR. Down syndrome, other full aneuploidies and polyploidy (En: Chromosome abnormalities and genetic counselling. Gardner RJM, Sutherland GR (eds.) Oxford university Press Inc., New York 1996, pp 252
- Martínez-Frías ML. Tabla 3.17: Síndromes cromosómicos (En: Defectos congénitos en España: Diez años de vigilancia epidemiológica. Dirección General de planificación Sanitaria, Ministerio de Sanidad y Consumo ed.) Secretaría General Técnica, Publicaciones, Madrid 1989, pp. 41 3. Jones KL. Trisomy 18 syndrome (En «Smith´s Recognizable Patterns of Human Malformation.
- Jones KL ed.) W.B. Saunders Co, Philadelphia1997, pp 16-17.
- Gardner RJM, Sutherland GR. Down syndrome, other full aneuploidies and polyploidy (En: Chromosome abnormalities and genetic counselling. Gardner RJM, Sutherland GR (eds.) Oxford university Press Inc., New York 1996, pp 252
- Martínez-Frías ML. Síndromes cromosómicos (En: Defectos congénitos en España: Diez años de vigilancia epidemiológica. Dirección General de planificación Sanitaria, Ministerio de Sanidad y Consumo ed.) Secretaría General Técnica, Publicaciones, Madrid 1989, pp. 41.