INFORMACIÓN BÁSICA.- Síndrome de Fraser (SF) (OMIM: 219000/ORPHA:2052/CIE-10:Q87.0). El síndrome de Fraser también conocido como síndrome de Meyer-Schwickerath, síndrome de Fraser-François, o síndrome de Ullrich-Feichtiger, es un trastorno congénito de herencia autosómica recesivo, ms frecuentes en fetos femeninos. La consanguinidad de los progenitores aparece en el 15-24,8% de los casos. No existe evidencia de transmisión vertical. No existe método preventivo, salvo el consejo genético.
El síndrome de Fraser es descrito por primera vez en 1962 por el genetista George R. Fraser.
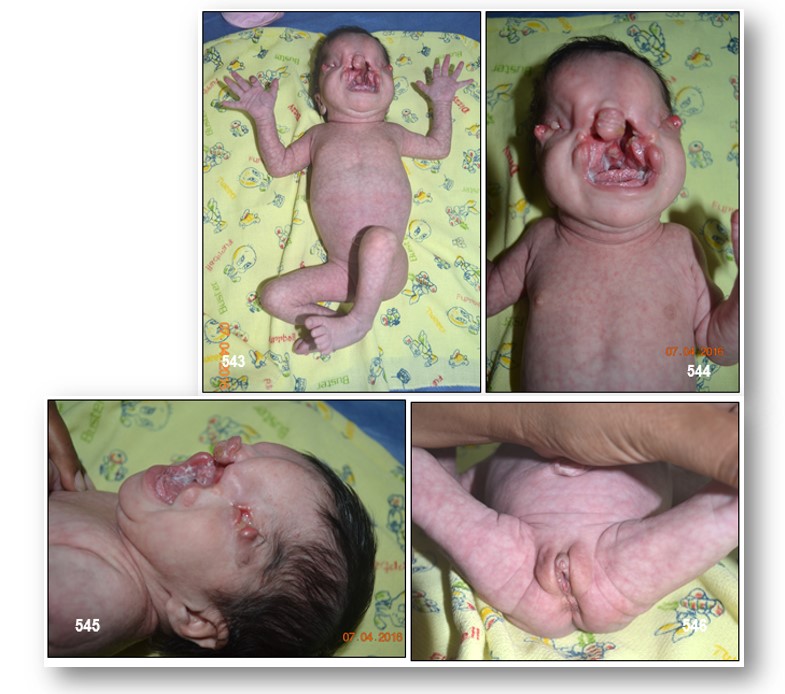
El criptoftalmos es la manifestación clínica más frecuente y consiste en la falta de formación de párpados, que deja la córnea y conjuntiva desprotegidas (de modo que estas estructuras pueden presentar una transformación dermoide). También puede cursar con alteraciones del segmento anterior y se pueden llegar a sustituir por tejido conectivo las estructuras del canal de Schlemm.
El diagnóstico precisa al menos 2 criterios mayores y uno menor, o bien un criterio mayor y cuatro menores (descritos por Thomas et al en 1986).
Epidemiología.- La incidencia del SF es de 0.043 por cada 10.000 niños nacidos vivos y 1,1 de cada 10.000 nacimientos sin vida, por lo que es un síndrome poco frecuente. En total se han descrito en la literatura alrededor de 150 pacientes afectados.
Genética Molecular.- El SF es una condición genéticamente heterogénea. Se han identificado mutaciones en el gen FRAS1 (cromosoma 4q21) y FREM2 (cromosoma 13q13). Ambos genes codifican proteínas de la matriz extracelular esenciales para la adhesión entre la membrana basal epidérmica y el tejido conectivo durante el periodo de la embriogénesis. Recientemente, se han descrito mutaciones en el gen GRIP1 (cromosoma 12q14) que codifica una proteína que interactúa con las proteínas anteriores, como causantes del SF. Mientras que se han descrito varias mutaciones en el gen FRAS1, solamente se han comunicado 2 mutaciones en el gen FREM2 hasta la fecha. Actualmente, se está estudiando el estado de portadores de la mutación en los padres.
En 5 familias con el síndrome de Fraser, McGregor et al. identificó 5 mutaciones homocigóticas en el gen FRAS1, que codifica una proteína de la matriz extracelular putativo.
En 2 familias con SF no vinculadas al gen FRAS1, Jadeja et al. encontrado una mutación homocigota en el gen FREM2.
En un lactante de sexo femenino con el síndrome de Fraser, Slavotinek et al. identificado heterocigosis compuesta para una eliminación y una inserción en el gen FRAS1, heredada de su madre y su padre, respectivamente.
Cavalcanti et al. denunció 2 hermanos varones brasileños nacidos muertos, natural de gestación 25 y 29 semanas, respectivamente. Una sib parecía tener una forma letal del síndrome de ablepharon-macrostomía o un fenotipo intermedio entre el AMS y el SF, y la otra tenía SF clásico. Análisis del gen FRAS1 reveló la homocigosidad para una mutación del sitio de empalme, lo que resulta en una proteína gravemente truncada en ambos hermanos y heterocigosidad para la mutación en ambos padres. Cavalcanti et al. llegó a la conclusión de que un fenotipo parecido AMS es una manifestación clínica poco frecuente de SF, sin genotipo/fenotipo correlación obvia.
En un feto femenino con un cariotipo normal y criptoftalmos, genitales externos ambiguos, sindactilia, pulmones bilobulado, agenesia renal bilateral, vejiga hipoplasia y agenesia de genitales internos con ovarios streak, Shafeghati et al. identificó homocigosidad para una mutación del sitio de empalme en el gen FREM2. Los padres iraníes consanguíneos fueron heterocigotos para la mutación. Un embarazo anterior había dado lugar a la muerte intrauterina a las 30 semanas de gestación de un feto de sexo masculino con un cariotipo normal en los que el diagnóstico del SF fue sugerido por la presencia de criptoftalmos, sindactilia, genitales ambiguos, ano imperforado, agenesia renal bilateral, pulmonar hipoplasia e hidrocefalia. Los autores señalaron que los hallazgos de los hermanos fueron consistentes con el SF clásico.
Entre las 18 familias consanguíneas con SF, Van Haelst et al. encontró 9 familias con vinculación a FRAS1, 3 familias a FREM2, y 3 familias de ambos genes. Seis familias no vinculan a cualquier lugar, lo que indica heterogeneidad genética.
Entre un grupo mayor de 33 familias, incluyendo las 18 familias consanguíneas, el análisis molecular identificó 11 nuevas mutaciones en el gen FRAS1 en 10 familias y 1 mutación en el gen FREM2 en 1 familia.
Una revisión de la literatura de genotipo/fenotipo correlaciones sugieren que los pacientes con FRAS1 mutaciones tienen mayor frecuencia de defectos de osificación del cráneo y una baja inserción del cordón umbilical en comparación con los pacientes sin una mutación FRAS1, pero los resultados no fueron estadísticamente significativos.
El ratón mutante «bl», un ratón generado en el laboratorio, tiene malformaciones similares a las del SF. De hecho, el ratón presenta una alteración en el gen equivalente al del hombre que causa la patología. De esta manera se podría estudiar mejor el origen del síndrome y cómo se producen las malformaciones.
Los resultados de este trabajo permitirán investigar algún tipo de tratamiento preventivo. Además, se podrá conocer mejor malformaciones como la agenesia/ displasia renal -desarrollo anormal de riñones-, frecuente en el SF, pero también en otros síndromes.
Mapping.- Por cartografía autocigotica, McGregor et al. situó el locus SF en el cromosoma 4q21. En 6 de 18 familias consanguíneas con SF, Van Haelst et al. excluídos vinculación tanto al FRAS1 y FREM2 genes, lo que indica heterogeneidad genética.
Manifestaciones clínicas.- El criptoftalmos, que etimológicamente significa ‘ojo oculto’, está presente en un 93% de los casos y se caracteriza por la ausencia de fisuras palpebrales, con pestañas ausentes y falta o malformación de los conductos lagrimales, lo que da un aspecto de parpados fusionados. Bajo los párpados fusionados suele haber un globo ocular poco desarrollado (microftalmía) o prácticamente ausente (anoftalmia).
La sindactilia (dedos de manos y pies fusionados) se manifiesta en un 54% de los casos. Son tambien frecuente las anomalías renales que por lo general suelen ser severas (riñones ausentes o multiquísticos), las malformaciones laríngeas (Atresia ó estenosis severa de laringe) y las genitales (fusión de labios y agrandamiento del clítoris, útero bicorne, malformación de trompas de Falopio en mujeres y testículos no descendidos y pene pequeño con hipospadias en varones). Otras malformaciones que pueden darse en el SF son: malformaciones del oído medio y la oreja; segmentación a lo largo del plano medio de las fosas nasales y la lengua; hipertelorismo; gran separación de la sínfisis púbica; desplazamiento de ombligo y pezones, y mesenterio común.
Diagnóstico.- El diagnóstico de este síndrome se puede hacer en el examen clínico y la autopsia perinatal. Koenig y Spranger observaron que las lesiones oculares son componentes aparentemente no obligatoria del síndrome. El diagnóstico del SF debe ser entretenido en pacientes con una combinación de malformaciones acrofacial y urogenital con o sin criptoftalmos. Thomas et al. También hizo hincapié en la aparición del síndrome de criptoftalmos sin criptoftalmos y propuso un criterio diagnóstico para el SF.
Los criterios principales consistieron en: criptoftalmos, sindactilia, genitales anormales, y la historia familiar positiva. Los criterios menores fueron: malformaciones congénitas de la nariz, las orejas o la laringe, labio leporino y/o paladar hendido, defectos esqueléticos, hernia umbilical, agenesia renal y retraso mental. El diagnóstico se basa en la presencia de al menos 2 principales y 1 criterios menores, o 1 criterio mayor y menor de 4. El labio leporino y / o paladar hendido, malformaciones cardíacas, anomalías músculo-esqueléticas y retraso mental se considera poco común. Van Haelst et al. (2007) sugirieron que el diagnóstico de SF se puede realizar si cualquiera de 3 criterios principales, 2 o mayor y 2 criterios menores, o 1 mayor y 3 criterios menores están presentes en un paciente.
Boyd et al. sugerido que el diagnóstico prenatal por ecografía de los ojos, dígitos y los riñones debe detectar la forma severa del síndrome. Serville et al. demostrado la viabilidad del diagnóstico ecográfico del SF a 18 semanas de gestación. Sugirieron que el diagnóstico se podría hacer si 2 de los siguientes síntomas están presentes: uropatía obstructiva, microftalmia, sindactilia y oligohidramnios. Schauer et al. hecho el diagnóstico de gestación 18,5 semanas «sobre la base de la ecografía. Tanto el feto femenino y el padre fenotípicamente normales tenían una anomalía cromosómica: inv. Un bebé nacido antes tenía el SF y el mismo cromosoma 9 inversión.
Van Haelst et al. prevista una revisión de los criterios de diagnóstico para el SF según Thomas et al. a través de la adición de las vías respiratorias y tracto anomalías del tracto urinario de los principales criterios y la eliminación de retraso mental y fisura como criterios. Los criterios principales incluyen sindactilia, espectro criptoftalmos, anomalías de las vías urinarias, genitales ambiguos, las anomalías de la laringe y la tráquea, y la historia familiar positiva.
Los criterios menores incluyen defectos anorrectales, orejas displásicas, defectos de osificación del cráneo, anomalías umbilicales, y anomalías nasales. El labio leporino y/o paladar hendido, malformaciones cardíacas, anomalías músculo-esqueléticas y retraso mental se considera poco común. Van Haelst et al. siguiere que el diagnóstico de SF se puede hacer si cualquiera de 3 criterios mayores o 2 mayores y 2 criterios menores, o 1 mayor y 3 criterios menores están presentes en un paciente.
Diagnóstico diferencial.- El diagnóstico diferencial incluye el síndrome de abléfaron-macrostomía, la displasia fronto-facio-nasal, el síndrome similar a Fraser, el síndrome de Meckel y la microftalmia sindrómica causada por mutaciones en heterocigosis del gen SOX2. También se debe considerar la displasia frontonasal y la criptoftalmia aislada.
Diagnóstico prenatal.- El diagnóstico genético preimplantacional o prenatal es posible si se han identificado mutaciones patogénicas responsables de la enfermedad en la familia. La ecografía prenatal puede detectar poli- u oligohidramnios, criptoftalmia, pulmones ecogénicos, agenesia o anomalías renales.
Conclusiones.- El caso descrito destaca fundamentalmente en dos aspectos. En primer lugar, cumple numerosos de los criterios diagnósticos para definirlo como SF (tres mayores y cuatro menores); pocos casos se aproximan tanto a los criterios definitorios. En segundo lugar, se trata de un síndrome muy infrecuente (sólo existen 117 casos publicados desde que Thomas et al describieron los criterios diagnósticos hasta el año 2002; si tenemos en cuenta series previas al establecimiento de los criterios diagnósticos, hay descritos cerca de 400 casos en la literatura científica).
Los criterios mayores (y su frecuencia) son: criptoftalmos (93-88%: 27,4% unilateral y 53% bilateral), sindactilia (54-79%), anomalías genitales (criptorquidia, hipospadias, pene hipoplásico, clitoromegalia, atresia vaginal, útero bicorne o rudimentario, genitales externos ambiguos…) y hermanos afectados. Los menores son alteraciones de nariz, oídos, laringe, paladar, hernia umbilical, agenesia renal uni o bilateral, anomalías esqueléticas y retraso mental.
Según Thomas et al y Koening and Spranger, el criptoftalmos es la malformación más importante para el diagnóstico de este síndrome, pero no una condición necesaria para el diagnóstico, y abogan por el término «síndrome de Fraser» y no por «síndrome de criptoftalmos».
Con respecto a los hallazgos clínicos del caso descrito, la incidencia de paladar ojival en los casos de SF es del 3,4% y la de hipoplasia nasal del 11,1%. La hipoplasia laríngea aparece en el 30,8% de casos y la estenosis subglótica e hipoplasia traqueal en el 8,5%. La hipertrofia de clítoris es la anomalía genital más frecuente en las mujeres con este síndrome (incidencia 36,8%) y sólo el 8% de casos presentan estenosis del introito vaginal. La ausencia de ovarios es muy infrecuente (tan sólo 2 casos de los 117 del estudio de Slavotinek tenían agenesia gonadal). La agenesia renal está presente en el 22,2% de casos. La mayoría de los casos descritos tienen cariotipo femenino normal, como sucede en nuestra paciente.
En 2003, Mc Gregor et al identificaron el gen responsable de la enfermedad como el 4q21 o FRAS 1, que codifica una proteína de matriz extracelular. Sus mutaciones se traducen en la finalización temprana de la síntesis de la proteína que codifica dicho gen. También se conocen otras mutaciones causantes de la enfermedad en otros genes distintos del FRAS 1.
No existen estudios endocrinológicos en estos pacientes, a pesar de la alta frecuencia de anomalías genitales. Las determinaciones realizadas en nuestro caso no tienen así referencias para realizar una comparación.
Pronóstico.- La supervivencia es variable y el pronóstico malo, por lo que el diagnóstico prenatal toma especial importancia. El 6% de casos son prematuros o abortos espontáneos, el 12% termina en interrupción voluntaria del embarazo y el 20% fallece en la primera semana de vida. El 13% sobrevive hasta 1 año de vida y otro 13% sobrevive entre 1 y 10 años. Las causas determinantes de la defunción son la estenosis/atresia laríngea, insuficiencia respiratoria, agenesia renal bilateral o una combinación de malformaciones renales y respiratorias. Los pacientes afectados que sobrevivieron más de 10 años apenas tenían malformaciones mayores y dudosamente cumplían con los criterios diagnósticos que hemos descrito. En otras series, el 50% sobrevive 1 año o más (aunque el 25% eran mortinatos). Los casos que sobreviven presentan retraso mental grave.
El caso que presento no tenía antecedentes familiares conocidos de malformados, mientras que el 41% de los casos tiene historia familiar de criptoftalmos o hallazgos compatibles con el SF.
No se objetivaron anomalías musculoesqueléticas (el 8-10% de los casos las presentan) ni gastrointestinales (7-12% de los casos). Aunque las malformaciones digestivas no son criterios diagnósticos, Slavotinek et al recomiendan que deberían considerarse, dada la frecuencia con que aparecen (atresia anal o anorrectal, estenosis anal, ano imperforado…).
Sólo el 18% de los casos de este síndrome presentan retraso de crecimiento.
Este caso destaca además por presentar alguna malformación extremadamente infrecuente como lo es el cutis marmorata.
Por último, el SF y el síndrome de Denys-Drash (DDS, OMIM 194080) son dos enfermedades superpuestas, que se caracteriza por el síndrome nefrótico asociado ya sea con la esclerosis difusa mesangial en DDS, o la esclerosis glomerular segmentaria focal en SF, defectos genitourinarios, y un mayor riesgo de desarrollar tumores, tales como tumor de Wilms en DDS, y el tumor gonadal tanto DDS y SF. Aunque no se observa con frecuencia en pacientes con SF, ha informado el tumor de Wilms.
Consejo genético.- El SF es una enfermedad autosómica recesiva. Se debe ofrecer consejo genético a las parejas de riesgo (ambos progenitores son portadores de una mutación causal de la enfermedad) informándoles que existe un riesgo del 25% de tener un hijo afecto en cada embarazo.
Bibliografía
- Fraser G.R. Our genetical load: a review of some aspects og genetical variation. Ann Hum Genet. 1962;25:387-415.
Zehender W. Eine Missgeburt mit hautueberwachsenen Augen oder Kryptophthalmus. Klin Monatsbl Augenheilkd. 1872;10:225-34.
Berg C, Geipel A, Germer U, Pertersen-Hausen A, Koch-Dörfler M, Gembruch U. Prenatal detection of Fraser syndrome without cryptophthalmos: case report and review of the literature. Ultrasound Obstet Gynecol. 2001;18:76-80. - Boyd PA, Keeling JW, Lindenbaum RH. Síndrome de Fraser (síndrome cryptophthalmos-syndactyly): revisión de once casos con hallazgos postmortem. Am J Med Genet. 1988 Sep; 31 (1): 159 – 168.
- Jadeja, S., Smyth, I., Pitera, JE, Taylor, MS, van Haelst, M., Bentley, E., McGregor, L., Hopkins, J., Chalepakis, G., Philip, N., Pérez Aytes, A., Watt, FM, Darling, SM, Jackson, I., Woolf, AS, Scambler, PJ identificación de un nuevo gen mutado en el síndrome de Fraser y blebs myelencephalic ratón. Nature Genet 37: 520 – 525, 2005.
- Shafeghati, A. Kniepert, G. Vakili, M. Zenker. Fraser syndrome due to homozygosity for a splice site mutation of FREM2. AmJ Med Genet Part A, 146A (2008), pp. 529-531
- McGregor, L., Makela, V., Darling, SM, Vrontou, S., Chalepakis, G., Roberts, C., Smart, N., Rutland, P., Prescott N. Hopkins J. Bentley, E., Shaw, A., y otros 13. El síndrome de Fraser y el fenotipo blebbed del ratón causado por mutaciones en FRAS1 / Fras1 que codifica una proteína putativa de la matriz extracelular. Naturaleza Genet. 34: 203 – 208, 2003.
- Serville, F., Carles, D., Broussin, síndrome de B. Fraser: detección prenatal de ultrasonidos. (Carta) Am. J. Med. Gineta. 32: 561 – 563, 1989.
- Van Haelst, MM, Maiburg, M., Baujat, G., Jadeja, S., Monti, E., Bland, E., Pearce, K., Síndrome de Fraser Collaboration Group, Hennekam, RC, Scambler, PJ estudio molecular de 33 familias con síndrome de Fraser: nuevos datos y revisión de mutaciones. A.m. J. Med. Gineta. 146A: 2252 – 2257, 2008.
- Van Haelst, MM, Scambler, PJ, Grupo de Colaboración del Síndrome de Fraser, Hennekam, RCM Síndrome de Fraser: estudio clínico de 59 casos y evaluación de criterios diagnósticos. A.m. J. Med. Gineta. 143A: 3194 – 3203, 2007.
- Cavalcanti, D. P, Matejas, V., Luquetti, D., Mello, M. F., Zenker, M. Fraser and ablepharon macrostomia phenotypes: concurrence in one family and association with mutated FRAS1. Am. J. Med. Genet. 143A: 241-247, 2007.
- McGregor, L., Makela, V., Darling, et al.: Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nature Genet. 34: 203-208, 2003.
- François J. Syndrome malformatif avec cryptophtalmie. Acta Genet Med (Roma). 1969;18:18-50.
Slavotinek AM, Tifft C.J. Fraser syndrome and cryptophthalmos: a review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genetics. 2002;39:623-33.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones M.C. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986; 25: 85-98.
Allali B, Hamdani M, Lamari H, Rais L, Benhaddou M, Kettani A, et al. Fraser syndrome. A case report. J Fr Ophtalmol. 2006;29:184-7.
Gattuso J, Patton MA, Baraitser M. The clínicasl spectrum of Fraser syndrome: report of three new cases and review. J Med Genet. 1987;24:549-55
Koening R, Spranger J. Cryptophthalmos B syndactyly syndrome without cryptophthalmos. Clin Genet. 1986;29:413-6.
Mc Gregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nature Genet. 2003;34:203-8.
Schauer GM, Dunn LK, Godmilow L, Eagle RC, Knisley A.C. Prenatal diagnosis and Fraser syndrome at 18.5 weeks gestation, with autopsy findings at 19 weeks. Am J Med Genet. 1990;37:583-91.
Fryns JP, Schoubroeck DV, Vandenberche K, Nagels H, Klerck P. Diagnostic echographic findings in cryptophthalmos syndrome (Fraser syndrome). Prenat Diagn.1997;17:582-4. - Fraser syndrome. Multiple congenital anomaly syndromes. Search Jablonskís Syndromes Database, 2006.