INFORMACIÓN BÁSICA.- Síndrome de Beckwith-Wiedemann (SBW). (OMIM: 130650). El SBW es un trastorno genético caracterizado por sobrecrecimiento, predisposición tumoral y malformaciones congénitas. Con una incidencia de uno entre 13.700 nacidos vivos, siendo mayor en niños engendrados con técnicas de reproducción asistida. No existe predilección por la raza ni por la edad. En cuanto a sexo, la incidencia es igual, salvo en los casos de gemelos monocigóticos en los que hay mayor incidencia en las mujeres. Uno de cada cinco casos fallece por complicaciones propias de la patología.
Las causas genéticas son muy complejas. La impronta genómica o imprinting es el fenómeno por el cual un gen se expresa de una manera o de otra según se herede del padre o de la madre. Alteraciones a nivel del cromosoma 11, particularmente de los genes que sufren imprinting, son los causantes del SBW. Aproximadamente el 50% de los casos se dan por alteraciones en la metilación. En la región 15.5 del brazo p del cromosoma 11 existen los genes CDKN1C, H19, IGF2 y el KCNQ1OT1, entre otros, implicados en el crecimiento. Las regiones del control del imprinting (ICR) se encargan de la metilación de estos genes. Una metilación anormal altera la regulación del crecimiento provocando la macrosomía. Entre 10-20% de los casos se da por una disomía uniparental paternal (dos copias paternas de los genes imprinting). Existen casos de mutaciones del gen CDKN1C, translocaciones o duplicaciones del material genético. Un 85% de los casos son esporádicos con un solo familiar afecto, en cambio en un 10-15% la herencia sigue un patrón autosómico dominante.
Existe un crecimiento aumentado durante la segunda mitad del embarazo, con fetos grandes para su edad gestacional. Algunos autores describen polihidramnios y placentomegalia. En los primeros años de vida presentan percentiles en torno al 97 en cuanto a talla y peso, y el perímetro craneal cercano al 50. Los percentiles comienzan a bajar alrededor de los 8 años, normalizándose en la edad adulta. Existen casos de hemihipertrofia. Es el síndrome pediátrico más común que implica hipercrecimiento.
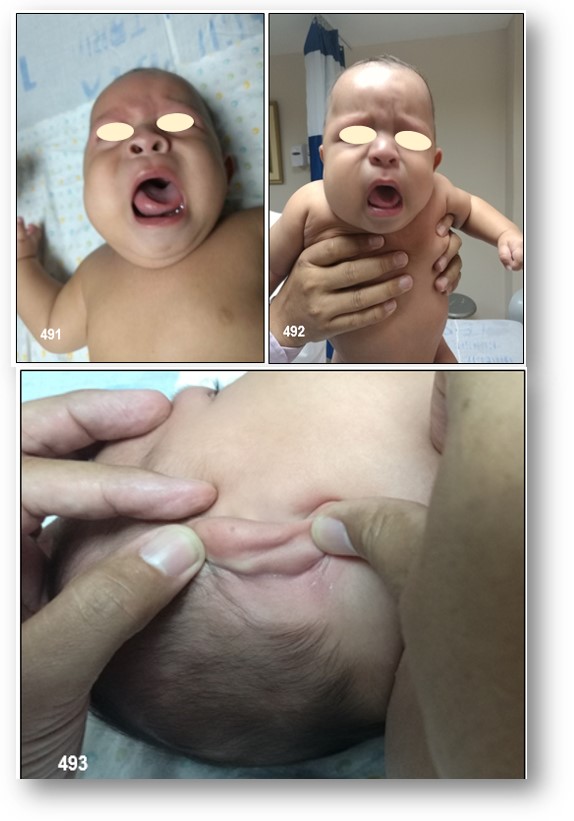
Son también frecuentes los defectos de la pared abdominal, tipo onfalocele, hernia umbilical y diástasis de los rectos, visceromegalia implicando uno o más órganos. La citomegalia adrenocortical fetal es patognomónica del SBW. La macroglosia es una de las malformaciones más frecuentes y puede originar graves repercusiones en el periodo neonatal. Pueden también presentar pliegues y surcos del lóbulo auricular, nevus flammeus facial u ojos prominentes. Otros hallazgos son malformaciones cardiacas. El patrón clínico es muy variable.
Existe una predisposición a tumores embrionarios, aumentado respecto a la normalidad en un 7%. Se suelen diagnosticar antes de los 8-10 años de vida. Los más comunes son el tumor de Wilms (50%) y el hepatoblastoma y, con menos frecuencia, rabdomiosarcoma, carcinoma adrenocortical y neuroblastomas. Existe un 30% más de riesgo de desarrollar tumores en los casos de pacientes con hemihipertrofia.
Elliot y cols. han establecido unos criterios diagnósticos para el síndrome (tabla 38). Implica la existencia de tres criterios mayores o dos mayores y tres criterios menores.

Es muy importante el diagnóstico en pacientes con menor expresividad fenotípica por el alto riesgo asociado a desarrollar tumores embrionarios y por las hipoglucemias. El seguimiento en el SBW consiste en cirugía de los defectos de la pared abdominal, screening de hipoglucemia y tratamiento si precisa, screening tumoral y somatometría. Pueden precisar cirugía reductora de la lengua por dificultad para la ingesta u obstrucción de la vía aérea. Ha de vigilarse la posibilidad de apnea del sueño. En ocasiones suelen necesitar el apoyo de logopedas y odontólogos.
El SBW tiene una baja incidencia que puede estar sesgada por la falta de diagnóstico en casos de menor expresividad clínica. Es una enfermedad que precisa de seguimiento cercano por su alta predisposición a tumores y por los casos de hipoglucemia.
Bibliografía
- Arroyo Carrera I, Martínez-Frías ML, Egüés Jimeno J, García Martínez MJ, Eloína Cimadevilla Sánchez C, Bermejo Sánchez E. Síndrome de Wiedemann-Beckwith: Análisis clínico-epidemiológico de una serie consecutiva de casos en España. An Esp Pediatr 1999;50:161-165.
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18(1):8-14.