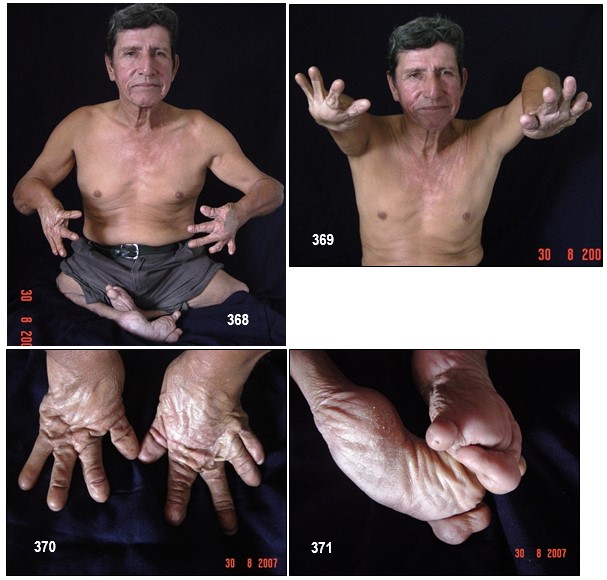
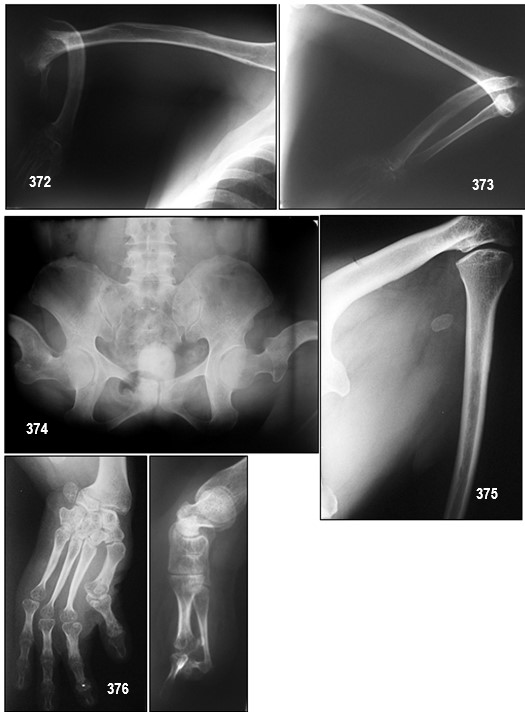
INFORMACIÓN BÁSICA.- Displasia mesomélica de Tipo Langer (DML).- (OMIM 249700).- Las displasias mesomélicas son un grupo heterogéneo de trastornos genéticos caracterizados por el acortamiento de la porción media de las extremidades. La DML fue clasificada por primera vez como una entidad única por Langer en 1967. Los sujetos afectados tienen extremidades cortas, hipoplasia de cúbito y peroné, hipoplasia mandibular de grado variable y habitualmente inteligencia normal.
Los padres de estos enfermos tienen, generalmente, talla baja y hallazgos clínicos y radiológicos de discondrosteosis de Leri-Weill.
La DML se caracteriza por una estatura desproporcionada y gravemente baja, con acortamiento mesomélico y rizomélico de las extremidades superiores e inferiores asociado con hipoplasia o aplasia de cúbito y peroné e hipoplasia mandibular. Pertenece al grupo de las displasias mesomélicas caracterizadas por acortamiento del segmento medio de los miembros. Este acortamiento es más marcado en pantorrillas que en antebrazos. Los pies y las manos son de morfología normal (hecho que no está presente en mi paciente). La desviación cubital de las manos es inconstante. La cara es de apariencia normal, salvo por la hipoplasia mandibular, que no está presente en todos los casos, y parece probable que no sea un componente genuino del síndrome. Hasta la fecha no se han descrito anomalías viscerales pero sí un caso de asociación con disgenesia gonadal. La inteligencia suele ser normal, aunque Robertson et al presentan un caso con retraso mental.
Los padres generalmente tienen talla baja y datos clínicos y radiológicos de discondrosteosis de Leri-Weill (DCLW). Ésta se caracteriza por talla baja moderada con acortamiento mesomélico de los miembros. Con frecuencia se asocia a deformidad de Madelung de la muñeca. Esta observación hizo surgir la hipótesis de que la DML es la expresión homocigota de la DCLW.
La prevalencia es desconocida, pero hasta el momento se han descrito menos de 50 casos en la literatura.
La DML representa la forma más grave de DCLW (con un acortamiento grave de los huesos largos de las extremidades, implicando al segmento medio y proximal), deformación de la cabeza humeral, angulación del eje radial, deformación del carpo, cuello femoral corto y ausencia o hipoplasia del segmento proximal de la tibia. En algunos casos se ha descrito hipoplasia maxilar leve. La deformidad de Madelung no es una manifestación típica de la DML, contrariamente a la DCLW. Las malformaciones asociadas ocurren raramente y la inteligencia es normal en casi todos los casos descritos. La DML se hereda de forma pseudoautosómica recesiva y está asociada a mutaciones homocigotas o heterocigotas compuestas y deleciones del gen SHOX (short stature homeobox; localizado en la región pseudoautosómica 1 (PAR1) de los cromosomas sexuales Xp22.33 y Yp11.32), o en la PAR1 downstream (dónde están localizados los elementos enhancer del SHOX). La DML forma parte de un espectro de enfermedades (que van desde las formas más graves como la DML, a formas más leves como la DCLW, la deformidad de Madelung aislada y la estatura corta idiopática), todas ellas asociadas a anomalías SHOX/PAR1. La prevalencia de las mutaciones SHOX/PAR1 se estima en 1/1000.
El diagnóstico de la DML puede sospecharse en base a los hallazgos clínicos y radiológicos y puede confirmarse por análisis moleculares (análisis de marcadores microsatélites, FISH o preferentemente, MLPA para las deleciones PAR1 y fusión de alta resolución (HRM), dHPLC y/o secuenciación de DNA para las mutaciones puntuales, deleciones pequeñas e inserciones de SHOX). La DML también se puede sospechar por ecografía en la semana 20 del embarazo y en esta etapa debe ser diferenciada del complejo fémur-fíbula-ulna (FFU) y de la forma Reinhardt–Pfeiffer de la displasia mesomélica. El consejo genético debe proponerse y se debe informar a las familias que las anomalías SHOX/PAR1 se heredan de manera pseudoautosómica dominante. Cada hijo de un individuo con DCLW tiene un 50% de posibilidades de heredar la mutación. Si ambos padres tienen DCLW, los descendientes tienen un 50% de posibilidades de tener DCLW, un 25% de posibilidades de tener DML y un 25% de no presentar ninguna de estas enfermedades. Todos los descendientes de un paciente afectado con DML y de un progenitor no afectado presentarán DCLW. El diagnóstico prenatal está disponible y aunque las solicitudes para la detección de estos trastornos son poco comunes, son más frecuentes para la DML que para la DCLW. No existe ningún tratamiento efectivo para la DML. El manejo médico sintomático de los niños con DML comienza en el nacimiento y continúa hasta la edad adulta. Es esencial un seguimiento exhaustivo del peso, altura y circunferencia de la cabeza. La baja estatura y las deformidades en las extremidades son graves, pero la esperanza de vida es normal.
La DML es, por tanto, una forma rara de enanismo, de herencia autosómica recesiva, en la que los padres están afectados de DCLW.
Las manifestaciones clínicas en los heterocigotos varían en intensidad, y muchas veces no son reconocidas, lo que impide realizar asesoramiento genético en estas familias. No obstante, es posible realizar diagnóstico prenatal al comienzo del segundo trimestre basándose en el desproporcionado acortamiento mesomélico de las extremidades y en la hipoplasia de cúbito y peroné.
Bibliografía
- Bueno M, Bueno-Lozano M, Bueno AL. Osteocondrodisplasias. En: Pombo M (ed). Tratado de Endocrinología Pediátrica, 2ª ed. Madrid: Díaz de Santos, 1997; págs. 331-348.
- Goldblatt J, Wallis C, Viljoen D, Beighton P. Heterozigous manifestations of Langer mesomelic dysplasia. Clin Genet, 31 (1987), pp. 19-24
- Langer LO. Mesomelic dwarfism of the hypoplastic ulna, fibula, mandible type. Radiology, 89 (1967), pp. 654-60
- Robertson SP, Shears DJ, Oei P, Winter RM, Scambler PJ, Aftimos S, et al. Homozygous deltion of SHOX in a mentally retarded male with Langer mesomelic dysplasia. J Med Genet, 37 (2000), pp. 959-64