INFORMACIÓN BÁSICA.- ENANISMOS: Síndrome de Laron, Enanismo tipo Laron o Síndrome de Insensibilidad a la Hormona del Crecimiento (OMIM 262500). – El Síndrome de Laron es una enfermedad endocrinológica hereditaria extremadamente rara que se caracteriza por la triada: dwarfismo (forma de enanismo con miembros bien proporcionados), rasgos faciales peculiares y niveles plasmáticos elevados de la hormona del crecimiento. Está causado por una anomalía en el gen que codifica la síntesis del receptor de la hormona de crecimiento (GHR) localizado en 5 p14-p12. Tiene una herencia autosómica recesiva, por alteraciones en homocigosis o heterocigosis compuesta. Estos pacientes presentan un profundo déficit de estatura que oscila entre -4 y -11.5 DE por debajo de la media, así como determinadas anomalías fenotípicas y riesgo de hipoglucemia.
Una condición clínica similar al Laron se ha relacionada con la participación de la insensibilidad a la GH posreceptor con un estado de inmunodeficiencia, se ha asociado con una mutación en el gen STAT5B (17q21.1) (OMIM 604.260).
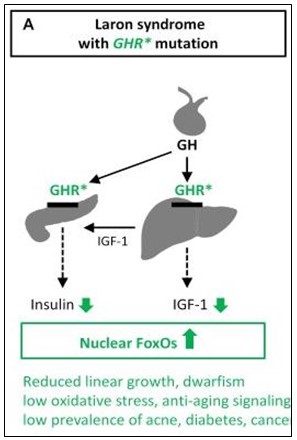
Esta condición genética que causa de enanismo y resistencia al cáncer.
Historia.- El síndrome lleva su nombre en honor a Zvi Laron, el investigador israelí que, junto con A. Pertzelan y S. Mannheimer. La resistencia a la GH fue por primera vez reportada por Laron en 1966, basado en observaciones comenzadas en 1958.
En el 2011, fue anunciado que el síndrome poseía un gen que proporcionaba resistencia al cáncer, a la diabetes y además del envejecimiento, ya que las células de los individuos que portan la mutación en el gen que codifica el receptor de la hormona de crecimiento activan un proceso de muerte celular controlada, o apoptosis (es una forma de muerte celular, que está regulada genéticamente), cuando se enfrentan a situaciones de estrés. Este aumento de la apoptosis da lugar a una menor acumulación de daño en el ADN de las células. De esta forma se limita la capacidad de estas células para entrar en procesos de transformación que dan lugar al cáncer.
Conocimientos fundamentales.- La AXIS GH (hormona de crecimiento) – IGF (factor de crecimiento similar a la insulina) tiene un papel bien establecido en la regulación del crecimiento somático, y hay muchas evidencias de que estas hormonas también contribuyen al crecimiento de los tejidos neoplásicos. Varios estudios experimentales y de población tratan de establecer una relación entre las concentraciones de IGF-I, IGFBP3 (proteína de unión a factor de crecimiento similar a la insulina) y el riesgo de cáncer.
Durante el período en que la hormona del crecimiento (derivados de glándulas pituitarias humanas) tenía un restringido a pacientes con discapacidades comprobadas indicó que ya existía una preocupación por la evaluación de los efectos adversos de este tratamiento.Con la introducción de la hormona recombinante (rhGH), las nuevas indicaciones terapéuticas, que ahora incluyen los pacientes con síndrome de Turner, insuficiencia renal crónica, retraso del crecimiento intrauterino, talla baja idiopática, síndrome de Prader-Willi surgió, además de la deficiencia de GH durante la edad adulta.También se ha cambiado el momento de uso, dosis y régimen de tratamiento.Debido al gran número de pacientes tratados, la preocupación acerca de los efectos adversos se hizo aún mayor, lo que lleva a la realización de varios estudios multicéntricos, la investigación y el consenso, estimulando aún más el debate sobre los posibles riesgos, tratando de crear las condiciones para que su uso caja de seguridad.
La primera evidencia de una posible asociación entre la GH y el cáncer surgió en 1950 con la demostración de que las dosis altas de GH, cuando se administra a ratones, causando cambios neoplásicos en varios órganos, mientras hipofisectomía revirtió un marco para la leucemia o protegido el animal el desarrollo de la enfermedad.
Más tarde, otros estudios han demostrado la actividad mitogénica de la GH y sus efectos tróficos sobre el tejido linfoide. En 1977, Rogers et al.planteado la hipótesis de la participación de la GH y somatomedina (IGF-I) en la patogénesis de la leucemia linfoblástica aguda en niños con leucemia después de detectar cambios de estas hormonas de acuerdo con la fase de la actividad o la remisión de la enfermedad.
Se han realizado varios estudios, lo que demuestra que la GH, debido a su metabolismo y el efecto mitogénico y IGF1, su efecto anti-apoptótica, actúan sobre la proliferación de las células normales y malignas a través del receptor de IGF-I, y son responsables de la inducción tumores en animales expuestos a dosis más altas de GH para inducir en la transformación in vitro de las células hematopoyéticas normales en las células leucémicas.
Estudios experimentales recientes indican, en primer lugar, que la GH puede producir tumores activos, lo cual sería una contraindicación para su uso, sin embargo, también se considera beneficioso en la reducción de la caquexia en pacientes con cáncer mediante la mejora de la supervivencia y justificando su indicación.
Cuando se analiza la relación entre el uso de rhGH y el aspecto de los tumores, surgen dos preguntas.La primera es la de rhGH indujo la aparición de tumores en pacientes sin enfermedad neoplásica, y es el segundo uno puede influir en la enfermedad recidiva tumoral en pacientes considerados para ser curado o en remisión.El propósito de este artículo es revisar las novedades de esta asociación, tanto desde el punto de vista fisiológico como clínica y epidemiológica.
Fisiología del sistema IGF.- El sistema incluye IGF IGF-I e IGF-II ligandos, receptores (tipo 1 y 2 de IGF, IGF-IR y el IGF-IIR, respectivamente) de las proteínas de unión a IGF (IGFBP1 6) y proteínas de señalización intracelular asociado con IGF-IR, que incluyen miembros de la IRS (receptor de insulina sustrato-) Akt, TOR (objetivo de rapamicina) y S6K (S6 cinasa) de la familia.
El hígado es responsable de la síntesis de mayor concentración de IGF y de IGFBP circulante, pero estas sustancias también puede ser secretada a nivel local en otros órganos por la acción autocrina o paracrina.GH es el principal estimulador de IGF-I, pero no de IGF-II.Las concentraciones de IGF-I se han incrementado sistemáticamente en paralelo con el postnatal y el crecimiento puberal, declinando después del cese de crecimiento y aumentó de nuevo en la vejez.IGF-II se expresa en altas concentraciones en embriones, pero después del nacimiento continúa para ser expresado y secretado por el hígado y está presente durante toda la vida.
Los IGFs son transportados a las células diana en el complejo con IGFBP, a diferencia de la insulina, lo que prolonga su vida media y modula su interacción con el receptor de membrana de la superficie. El IGFBP3 entre las IGFBPs, es la más abundante forma circulante, responsable de la mayor parte de la capacidad de unión a IGF, en particular, el IGF-I; y por lo general se asocia con un complejo ternario que consiste en ella, el IGF-I y ALS (subunidad lábil en medio ácido).El IGFBP1 y IGFBP6 tener una capacidad de 10 veces más alto de unión a IGF-II sobre el IGF-I, mientras que las otras IGFBPs tienen igual afinidad por los IGF.Ninguna de las IGFBPs se une específicamente a la insulina.
Las principales diferencias entre los IGF e insulina están en la estructura, los receptores preferidos, interacciones con proteínas de unión, y en consecuencia la función. La insulina actúa como un aglutinante para suelta activar su receptor (IR), mientras que el IGF-I y II, una vez unido a las IGFBP, active principalmente de IGF-IR, que es similar a la estructura de IR. IGF e insulina, aunque a un menor afinidad últimos tienen la capacidad de unirse a un receptor híbrido IGF-IR e IR. IGF-II también se une a una isoforma relacionada Una-IR (insulina relacionada con IR-A) y el receptor de IGF-IIR (también llamado receptor de 6-fosfato de IGF-II/manose).La función principal de la IGF-IIR es ser un regulador negativo de la biodisponibilidad de IGF-II.
Cualquier receptor del IGF-IR e IR dominio membranosa parte como de feria intracelular C-terminal tiene un número menor de secuencias idénticas que el dominio de tirosina quinasa, que confiere especificidad. Después de la unión de IGF-I o IGF-II de la actividad de IGF-IR del dominio de tirosina quinasa catalítica que desencadena la fosforilación de sustratos intracelulares que se producen (Figura 1).
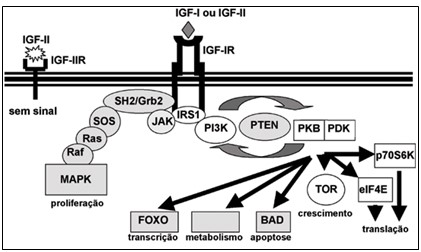
La proteína IRS (sustrato del La proteína IRS (sustrato del receptor de insulina) 1 a 4 y en situación especialmente difícil (colágeno homología Src) son la mayoría de los sustratos para la fosforilación intracelular. El fosforilados IRS recluta de PI3K (fosfatidilinositol 3-quinasa), resulta en la activación de PDK (fosfoinositida-dependiente de la proteína quinasa), PKB (proteína quinasa B) y p70S6 quinasa cascada que controlan la transcripción a través de FOXO (factores de transcripción forkhead), el metabolismo a través de GSK-3b (glucógeno sintetasa quinasa 3 b), la apoptosis a través de BAD (promotor de la muerte asociada a Bcl), el crecimiento celular a través de TOR y la traducción a través de eIF4E (factor 4E iniciación eukaryothic)-S6K. La activación de la proliferación celular está mediada por la unión de la proteína adaptadora Grb2 SH2 (receptor del factor de crecimiento de la proteína unida 2) receptor, que desencadena la activación de SOS, Ras, Raf (proteínas GTPasa mediada por la familia) y MAPK (mitogen-activated proteína quinasa). Reguladores negativos de este sistema incluyen la Grb2, el sistema JAK (Janus quinasa de tirosina) y PTEN (fosfatasa mutado en el cromosoma 10) ( Figura 1). La unión de IGF-II con su respectivo receptor (IGF-IIR) no desencadena señales intracelulares por no tener el dominio de tirosina quinasa, y parece tener acción negativa sobre la proliferación mediante la reducción de las cantidades de IGF-II de unión a IGF-viables- IR.
Fisiopatología/Genética. – Investigaciones de genética molecular han demostrado que este trastorno se asocia principalmente con las mutaciones en el gen que codifica para el receptor de GH. Estos pueden resultar en defectuosa unión de la hormona a la ectodominio o reducción de la eficiencia de la dimerización del receptor después de la ocupación de la hormona.
Hay niveles excepcionalmente bajos de factor de crecimiento similar a la insulina (IGF-1) y su proteína portadora principal de crecimiento tipo insulina proteína de unión al factor de 3 (IGF BP-3).
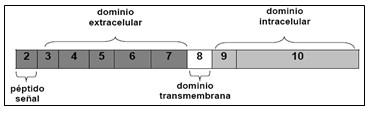
- Gen en el cromosoma 5 de humanos
- Con 9 exones:
- Exón 2 codifica el péptido señal
- Exones 3 – 7 codifican dominio extracelular
- Exón 8 codifica el dominio transmenbrana
- Exones 9 – 10 codifican el dominio intracelular, además de codificar la GHBP
GHR:
- Es el receptor en su forma completa
- De la clase I de las citoquinas/hematopoyetinas
- Región BOX1: secuencia rica en prolina esencial para la transducción
- Dimerización de heterodímeros u homodímeros para la transducción de señal.
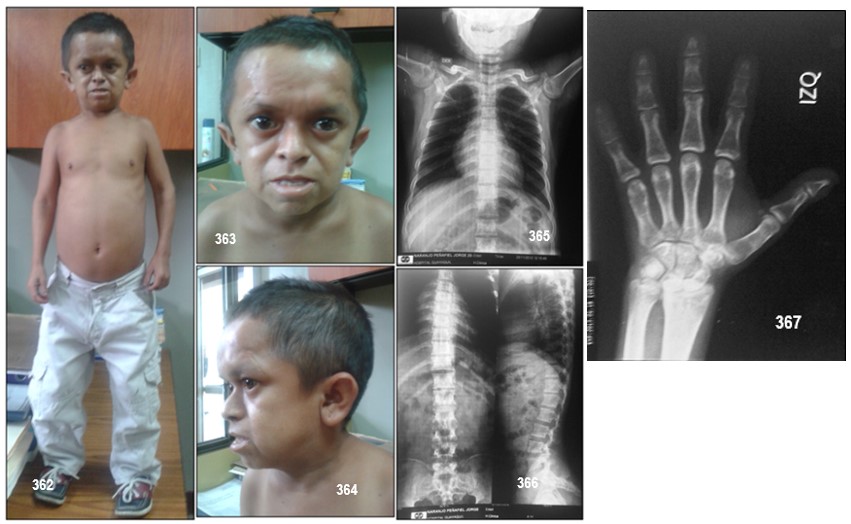
Síntomas.- Clínicamente los niños de talla y peso normales al nacimiento, tienen rasgos faciales característicos: cabeza pequeña y redondeada, cara corta y ancha, hueso frontal prominente, nariz pequeña, puente nasal deprimido y en forma de silla de montar, pliegues nasolabiales bien desarrollados, mandíbula y mentón hipoplásicos (hipoplasia es el desarrollo incompleto o defectuoso), exoftalmos (protrusión anormal del globo del ojo); los dientes brotan de forma tardía y suelen estar apiñados; cuando alcanzan la edad adulta presentan además dwarfismo con acromicria (anomalía caracterizada por la existencia de manos y pies anormalmente pequeños). Se acompaña de cuello corto y laringe pequeña, voz de tono agudo después de la pubertad; genitales hipoplásicos con maduración sexual retrasada o que no llega a producirse, los caracteres sexuales secundarios, vello facial, vello axilar y vello pubiano suelen estar ausentes; el cabello es fino y la inteligencia suele ser normal.
La mayoría de los casos han sido reportados en poblaciones con ascendencia semita o árabe en países como Israel, Arabia Saudita, Egipto e Irak. Caso aparte es una villa en la Ciudad de Loja, Ecuador, en donde existe una gran población con el mismo mal.
Diagnóstico.- El diagnóstico de sospecha es clínico y el diagnóstico de confirmación se realiza mediante estudio hormonal. Aunque los enfermos producen niveles normales de hormona del crecimiento, sus niveles en sangre están muy elevados porque el organismo no sabe utilizar la hormona correctamente; presentan bajas concentraciones de IGF-1 circulantes. Tienen una respuesta normal o alta a la hormona del crecimiento ante las pruebas provocadoras y respuesta baja o nula de la IGF-1 frente a las inyecciones de la hormona del crecimiento. La radiografía lateral de cráneo y la resonancia magnética nuclear cerebral permiten descartar patología hipotálamo-hipofisaria y la radiografía de mano y muñeca sirven para evaluar la maduración ósea y como pronóstico de la talla final aproximada.
Para establecer este diagnóstico, se realizan dosajes séricos de hormona de crecimiento (GH) e IGF-1 tanto basales como estimulados, evidenciándose el incremento de GH y disminución de IGF-1, de la misma manera del transportador IGF BP3, hallazgos hormonales propios del síndrome de Laron. La confirmación diagnóstica se establece mediante un estudio secuencial del DNA del gen que codifica para el receptor GH (GHR), y que la gran mayoría presenta un compromiso en el dominio citoplasmático. Aunque se han descrito otras alteraciones como defectos en el EXON 8 (splice mutations).
Tratamiento.- La administración de GH no tiene efecto sobre la producción de IGF-1, por lo tanto el tratamiento se basa principalmente en dar dosis directas del IGF-1. IPLEX® se compone de recombinante humano de IGF-1 y su proteína de unión a IGFBP-3. Fue aprobado por la Administración de Drogas y Alimentos de EE.UU. en 2005 para el tratamiento de la deficiencia primaria de IGF-1 o GH deleción del gen. Los efectos secundarios de IPLEX son hipoglucemia. Empresa de fabricación de IPLEX®, Insmed, ya no se puede desarrollar proteínas y ya no puede fabricar IPLEX® como de un comunicado dado a conocer en enero de 2012.
Bibliografía
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=633
- Laron Z, Pertzelan A, Mannheimer S (1966). «Genetic pituitary dwarfism with high serum concentation of growth hormone–a new inborn error of metabolism?». Isr. J. Med. Sci. 2 (2): 152-5.
- Laron Z (2004). «Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003». J. Clin. Endocrinol. Metab. 89 (3): 1031-44.
- Laron Z, Ginsberg S, Lilos P, Arbiv M, Vaisman N (2006). «Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity)». Clin. Endocrinol. (Oxf) 65 (1): 114-7.
- «Pueblo ecuatoriano con enanismo porta gen que evade al cáncer y la diabetes».