INFORMACION BASICA: MUCOPOLISACARIDOSIS: Mucopolisacaridosis (MPS).-(ORPHA:79213).– Son un grupo de enfermedades originadas en errores innatos del metabolismo de los glucosaminoglicanos* (GAGs), anteriormente llamados mucopolisacáridos, que producen acumulación progresiva de estas moléculas en los lisosomas de las células del tejido conectivo, incluido cartílago y hueso. Son causadas por la deficiencia de las enzimas lisosomales que los degradan. Las enzimas lisosomales rompen las largas cadenas de polisacáridos en unidades menores dentro del lisosoma. Los fragmentos resultantes son nuevamente degradados por hidrólisis secuenciales de sus terminaciones; las
hidrolasas que intervienen en este proceso son 10 y su deficiencia produce depósito intralisosomal de glucosaminoglicanos incompletamente degradados, los cuales son almacenados en el citoplasma alterando la fisiología celular. La ocurrencia conjunta en países europeos se estima de 1:10.000 a 1:25.000 recién nacidos vivos; es más frecuéntela MPS de tipo III. La ocurrencia en Australia es 1:107.000 nacidos vivos para síndrome de Hurler, 1:320.000 para síndrome de Hunter, 1:58.000 para síndrome de Sanfilippo, 1:640.000 para síndrome de Morquio y 1:320.000 para síndrome de Marotaux-Lamy.
Los GAGs sirven para formar la pared y matriz celular, son producidos por todas las células, participan en la formación estructural y comunicación celular, se los encuentra fundamentalmente en el tejido conectivo, epitelios y óseo.
Epidemiología: En nuestro país, y en muchos países del continente, es difícil el cálculo de la frecuencia de estas enfermedades, porque generalmente solo se diagnostican algunos casos que usualmente corresponden a los casos más graves, siendo poco diagnosticados los casos “atenuados”.
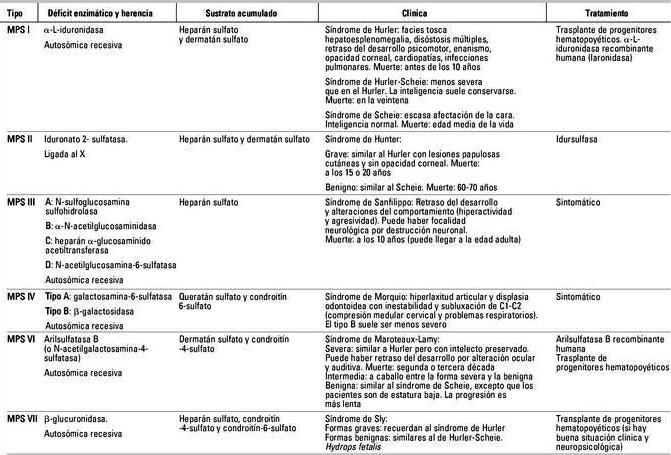
Clasificación: Se basa en el defecto enzimático (11 tipos diferentes) y los fenotipos característicos (9). Un fenotipo puede ser causado por más de un desorden enzimático.
En el caso de la enfermedad de Morquio hay 2 defectos enzimáticos diferentes, en el síndrome de Sanfilippo. Para un mismo defecto enzimático pueden existir diversos grados de gravedad en el espectro clínico: en la MPS I, el síndrome de Hurler es la forma más grave, con afectación del sistema nervioso central; el síndrome de Scheie es la forma menos grave, y hay una forma intermedia (Hurler-Schie). La clasificación actual comprende siete tipos de MPS: I, II, III, IV, VI, VII y IX (véase la tabla 22).
Genética: En general, todas son autosómicas recesivas salvo dos excepciones, las enfermedades de Hunter, que son ligadas con el cromosoma X. Se conocen muchos de los genes que codifican estas enzimas y se han establecido correlaciones genotipo-fenotipo para algunas de ellas, aunque la gran variabilidad fenotípica indica un alto índice de mutaciones. La mutación más frecuente en Rusia y Escandinavia es Q70X. Por otro lado, algunos tipos de MPS son causados por más de una mutación; en el caso de la enfermedad de Hurler.
Patogénesis: Las enfermedades de depósito lisosomal son un grupo de enfermedades en las que se encuentran defectos en múltiples niveles de la síntesis y degradación de las hidrolasas lisosomales: alteraciones de la síntesis y plegamiento, defectos de la activación, defectos en los sustratos y defectos en las proteínas de membrana.
Según el concepto expresado, la continua captación lisosomal de material no metabolizado produce hipertrofia de los lisosomas con alteración de la función celular y posible destrucción de las células afectadas. Los signos y síntomas de cada una de las enfermedades reflejan, en general, el patrón de distribución de los productos naturales no degradados a causa de la deficiencia enzimática, pero esta acumulación dentro de los macrófagos no explica del todo el crecimiento visceral ni muchas de las manifestaciones. Por ejemplo, en la enfermedad de Fabry la hipertrofia cardíaca puede llevar a gran crecimiento cardíaco, pero menos de 0,5% es material de depósito. La acumulación del material no degradado resulta en alteraciones estructurales visibles con microscopios de luz y electrónico. En algunas enfermedades la acumulación del material de depósito lleva al aumento de citoquinas o al aumento intracelular de óxido nítrico, lo que va a alterar la función del macrófago.
En los pacientes con afectación neurológica también se encuentra activación de los macrófagos, que es una de las razones por las cuales hay daño cerebral. Otra alteración notoria es el aumento del calcio intracelular, que va a contribuir a la muerte neuronal, por ejemplo en la enfermedad de Gaucher. La acumulación extralisosomal del material de depósito produce alteraciones de la señalización transmembrana e intracelular, lo que origina alteraciones de la función mitocondrial.
Los eventos secundarios a la acumulación del material de depósito pueden no ser susceptibles de mejorar con el tratamiento y esto debe ser tenido en cuenta.
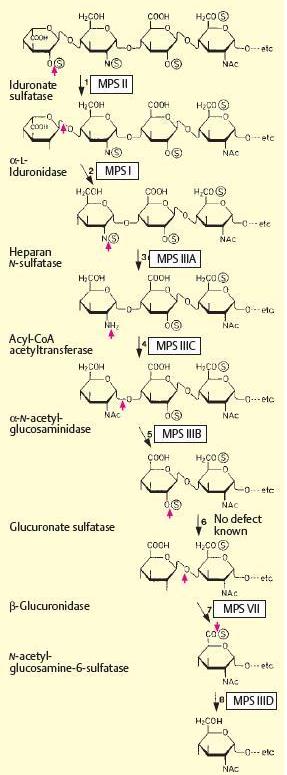
La mayoría de estas enfermedades se deben a una alteración enzimática, pero pueden ser causadas por la alteración de más de una vía enzimática; por ejemplo, la enfermedad de Morquio tiene dos defectos enzimáticos y le enfermedad de Sanfilippo cuatro alteraciones.
La gravedad de estas enfermedades está relacionada con la actividad residual de las enzimas; se considera que existe un umbral para esta actividad residual, por debajo del cual viene la incapacidad para manejar el sustrato y la consiguiente acumulación. Se ha demostrado que cambios pequeños en esta actividad residual puede ocasionar alteraciones serias en la tasa de acumulación del sustrato.
En general, se considera que a menor actividad residual de la enzima más tempranamente se iniciarán las manifestaciones clínicas y por ende su gravedad (en enfermedad de Gaucher es un poco diferente). En aquellas enfermedades para las cuales hay terapia de reemplazo enzimático disponible, mientras menor actividad residual menos respuesta clínica se obtendrá.
Las manifestaciones clínicas no se dan únicamente por el acúmulo del material depositado, sino por interferencias intracelulares e intercelulares, especialmente en los mecanismos de señalización.
Manifestaciones Clínicas: Son enfermedades de curso crónico, progresivo, con gran variabilidad en la gravedad y evolución de los síntomas. La acumulación excesiva de glucosaminoglicanos en los tejidos hace que los pacientes afectados tengan un fenotipo dismórfico con facies característica (véase la imagen/paciente con probable síndrome de Morquio) y afectación multisistémica, principalmente esquelética y visceral. Cada tipo de glucosaminoglicano tiene órganos de depósito principales: el heparán sulfato produce síntomas predominantemente neurológicos, como en las enfermedades de Hurler y Hunter y el síndrome Sanfilippo; el keratán sulfato produce opacidades corneales y alteraciones esqueléticas sin afectación neurológica, como en el síndrome de Morquio y el dermatán sulfato produce miocardiopatía y valvulopatías, como en las MPS I, II y VI. Fenotipo característico (dismorfismo).
La facies típica de los pacientes con MPS es llamada también gargolismo o facies hurleriana, en remembranza de las primeras descripciones de la enfermedad, la cual a medida que progresa la hace más evidente. Consiste en aspecto tosco, escafocefalia, prominencia frontal, pelo y cejas gruesos, puente nasal bajo, lengua protruyente y cara ancha por engrosamiento de los huesos faciales. Los pacientes pueden nacer con talla normal y permanecer con percentiles de talla normales hasta el primer año de vida, posterior al cual hay detención de la rata de crecimiento, principalmente relacionada con las alteraciones óseas (displasia esquelética), que son múltiples y progresivas; véase la Tabla 2.
Diagnóstico: Prenatal. Puede hacerse por estudio de amniocitos o de vellosidades coriónicas; en el período neonatal se pueden hacer pruebas de tamización cualitativas para la determinación de glucosaminoglicanos en orina, teniendo en cuenta que estas sustancias acumuladas en los lisosomas son excretados en la orina de los afectados. Cuando se sospecha una MPS se utilizan pruebas como albúmina ácida, cloruro de cetilpiridinio, tests cuantitativos como: el azul de 1,9-dimetilmetileno, cromatografía en capa fina o electroforesis para identificar el tipo de glucosaminoglicano excesivamente excretado. El diagnóstico confirmatorio se hace con estudio de la actividad enzimática en plasma, leucocitos o fibroblastos. Hay una técnica para medir la actividad de algunas enzimas lisosomales en sangre total recolectada en papel de filtro. Actualmente se ha descrito la técnica de azul de 1,9-dimetilmetileno en orina seca en papel de filtro, especialmente en MPS I. En nuestro país, lamentablemente no se puede hacer tamización cualitativa y cuantitativa neonatal en orina y sangre, con confirmación del déficit enzimático específico en muestras enviadas en papel de filtro a un laboratorio de referencia que determinará la actividad enzimática. Para la interpretación de los exámenes paraclínicos debe tenerse en cuenta que la excreción de los glucosaminoglicanos disminuye con la edad; además, las MPS III y IV pueden no ser fácilmente detectadas debido a la baja excreción de glucosaminoglicanos en la orina de los pacientes afectados, por lo que se recomienda realizar el test de azul de 1,9-dimetilmetileno. Se han identificado los genes responsables de la mayoría de las MPS, excepto la MPS III-C.
Tratamiento: Sintomático. Se está ensayando terapia de reemplazo enzimático. La terapia de remplazo, la plasmaféresis y el trasplante de fibroblastos no han demostrado utilidad en el tratamiento de los pacientes con MPS. El trasplante de médula ósea es útil en caso de enfermedades de Hurler y de Maroteaux-Lamy antes de los 2 años de vida: disminuye la afectación facial, visceral, rigidez articular, deterioro cardíaco, deterioro cognoscitivo, obstrucción de la vía aérea, pero no mejora las alteraciones óseas. Es posible diagnosticar las portadoras mediante medición de la actividad enzimática en MPS tipo II, cuya herencia es recesiva ligada con el cromosoma X, al igual que hacer el diagnóstico prenatal mediante la medición de la actividad enzimática en las vellosidades coriónicas y cultivo de células de líquido amniótico.
Luego de estas generalidades, me réferi en detalles a la MPS tipo IV (Enfermedad de Morquio) presentado en el texto.
MUCOPOLISACARIDOSIS TIPO I O ENFERMEDAD DE HURLER
INFORMACIÓN BÁSICA.- Mucopolisacaridosis Tipo I. Síndrome de Hurler (OMIM 607015). Es una rara enfermedad hereditaria del metabolismo de los glucosaminoglicanos (antes denominados mucopolisacáridos). Otros síndromes de almacenamiento lisosomal, se descubrieron en la década del sesenta, cuando se identificaron los glucosaminoglicanos dermatán y heparán sulfato en la orina, en pacientes con síndromes de Hurler, Scheie y Hunter; heparán sulfato en el síndrome de Sanfilippo; queratán sulfato y condroitín sulfato en el síndrome de Morquio y dermatán sulfato en el síndrome de Marotaux-Lamy.
Herencia.- Se presenta un caso por cada 100.000 nacimientos. El síndrome de Hurler es hereditario, autosómico recesivo, lo cual significa que se transmite de padres a hijos. Ambos padres necesitan transmitir el gen defectuoso (heterocigotos al gen) para que su hijo desarrolle este síndrome. El síndrome de Hurler pertenece a un grupo de enfermedades llamadas mucopolisacaridosis o MPS.
Nombres alternativos.- Deficiencia de alfa-L-iduronidasa; Mucopolisacaridosis tipo I; MPS 1 H
Causas.- Sin esta enzima, la alfa-L-iduronidasa, los glucosaminoglicanos se acumulan y causan daño a órganos, por el depósito progresivo de mucopolisacaridos en las células del tejido conectivo, incluido cartílago y hueso. Es una enfermedad por depósito o tesaurismosis. Este síndrome es el tipo más severo y está categorizado como MPS I H. Los otros subtipos de MPS I son: MPS I HS (síndrome de Hurler-Scheie); MPS I S (síndrome de Scheie)
Síntomas.- Los síntomas pueden ir de leves a severos y generalmente aparecen entre 3 y 8 años de edad. Los bebés con el síndrome de Hurler severo parecen normales al nacer y los síntomas faciales se pueden hacer más notorios durante los primeros dos años de vida. Los síntomas abarcan: Huesos anormales en la columna; Mano en garra; Córneas opacas; Sordera; Crecimiento interrumpido; Problemas cardíacos; Enfermedad articular incluyendo rigidez: Discapacidad intelectual que empeora con el tiempo; Rasgos faciales gruesos y toscos con puente nasal bajo.
Los individuos con la variante clásica del Síndrome de Hurler tienen una apariencia similar. A menudo tienen la cara regordeta con las mejillas rosadas y la cabeza bastante grande con una frente prominente. El cuello es corto y la nariz ancha con un puente aplanado (silla de montar) y los orificios nasales antevertidos. Las órbitas de los ojos son poco profundas y los ojos sobresalen.
Los labios son a menudo gruesos y la lengua grande. El pelo tiende a ser espeso (hirsuto), las cejas espesas y puede haber más pelo de lo normal en el cuerpo. Tienen el abdomen prominente y una manera muy característica de andar y sostener los brazos debido a contracturas en las articulaciones de las caderas, los hombros, los codos y rodillas.
La apariencia de los individuos con la forma atenuada o Scheie puede variar. Los niños pueden parecerse a los niños sanos. Los adultos son normalmente más bajos que la mediana, tienen una manera característica de caminar debido a que sufren contracturas en las articulaciones y tienen el tronco a menudo más corto que las extremidades. El cuello es corto, los labios pueden ser gruesos y la mandíbula cuadrada. Otros pueden adquirir en el futuro los mismos rasgos físicos de los afectados por la variante clásica del Hurler pero la progresión es más lenta.
En la variante clásica de MPS I la inteligencia queda afectada y al final los niños acaban perdiendo las habilidades ya aprendidas.
Los afectados varían mucho incluso en la forma severa de MPS I; mientras algunos niños llegan a decir sólo unas cuantas palabras, otros aprenden a hablar bien y a leer un poco. Los padres dan énfasis a que es importante ayudar a los bebés con Hurler a aprender tanto como sea posible antes de que la enfermedad progrese.
Incluso cuando el niño empieza a perder las habilidades que ha aprendido puede que nos sorprenda con cosas que aún conserva. Pueden seguir comprendiendo y disfrutar de la vida aún cuando hayan perdido la habilidad de hablar.
No hay almacenamiento de mucopolisacáridos en el cerebro de los afectados por la variante Scheie y generalmente tienen una inteligencia normal. Otros que no tienen la enfermedad de Hurler clásica pueden ser de inteligencia normal, pero algunos tendrán dificultades moderadas de aprendizaje.
En algunos individuos con la forma clásica de MPS I la circulación del líquido cefaloraquídeo puede bloquearse a medida que avanza la enfermedad.
En caso de convulsiones y/o crisis epilépticas se realiza un EEG y el neuropediatra o neurólogo les aconsejará si precisa tratamiento antiepiléptico.
A nivel de los ojos, la opacidad corneal causada por el depósito de mucopolisacáridos se puede observar en todos los niños y adultos con MPS I y puede llevar a una discapacidad visual significativa. La opacidad corneal severa puede reducir la vista, sobre todo con poca luz. Algunos individuos no toleran las luces brillantes ya que la opacidad provoca una refracción desigual de luz. Algunos pacientes precisan de un trasplante de córnea.
Es común tener algún grado de sordera en individuos que padecen los diversos tipos de MPS I. Puede ser sordera de conducción del nervio o ambas (sordera mixta) y posiblemente empeorada por infecciones frecuentes de oído.
Es importante que a los afectados por MPS I se les haga un control auditivo de forma regular para que los problemas puedan tratarse lo antes posible y así mejorar o mantener la habilidad de comunicarse y aprender lo antes posible. La sordera de conducción tiene que ver con la transmisión deteriorada de las ondas sonoras a través del canal auditivo, el tímpano y el oído medio. El correcto funcionamiento del oído medio depende de que la presión detrás del tímpano sea la misma que la del canal auditivo exterior y la de la atmósfera.
La presión la iguala la trompa de Eustaquio que va del oído medio a la parte trasera de la nariz. Si la trompa se bloquea la presión detrás del tímpano baja y éste puede quedar afectado. El resultado de todo esto es un deterioro en la transmisión de las ondas sonoras. Si la presión incorrecta persiste el líquido del revestimiento del oído medio se acumulará y con el tiempo se volverá tan viscoso como el pegamento. Por eso esta afección se conoce con el nombre de “oído de pegamento o cera”.
Los problemas descritos en este apartado son comunes en los niños con la variante clásica de Hurler y en menor grado en aquéllos con Hurler Scheie. Los que tienen el síndrome de Scheie suelen estar menos afectados.
Típicamente el puente de la nariz es aplanado y el paso de detrás de la nariz es menor debido al pobre crecimiento de los huesos de la cara y la textura espesa de la cobertura mucosa.
La combinación de huesos anormales y almacenamiento de mucopolisacáridos en los tejidos no rígidos de la nariz y la garganta puede causar fácilmente una obstrucción en la nariz. Uno de los rasgos comunes de niños con la enfermedad de Hurler es la descarga continua de mucosidad clara por la nariz y las infecciones de los senos nasales.
Tos frecuente, resfriados e infecciones de la garganta son problemas comunes en los afectados por MPS I. A menudo las amígdalas y vegetaciones adenoideas se agrandan y pueden bloquear parcialmente las vías respiratorias.
Por esta razón se pueden operar (vea dificultades respiratorias). El cuello es normalmente corto lo que favorece la existencia de problemas respiratorios. La tráquea se estrecha por la acumulación de mucopolisacáridos.
Los labios son más gruesos, las encías anchas y la lengua grande. Los dientes están más distanciados, mal formados y con el esmalte frágil. Es importante que los dientes se cuiden bien para evitar que las caries puedan causar dolor y la pérdida de piezas dentales.
En la variante clásica de Hurler la forma del pecho es anormal y la unión entre las costillas y el esternón no es tan flexible como debería ser. Las costillas son anormalmente rectas y tienen la forma de remos con el cuello estrecho y las puntas anchas por lo tanto el pecho es rígido e incapaz de moverse libremente para permitir que los pulmones inspiren un gran volumen de aire. Los nódulos o la excesiva dureza de los tejidos pueden bloquear las vías respiratorias.
El hígado y el bazo son más grandes de lo normal empujan hacia arriba los músculos de la base del pecho (el diafragma), reduciendo así aún más el espacio para los pulmones. Cuando los pulmones no se limpian totalmente, hay un mayor riesgo de infección (neumonías).
El tejido pulmonar se espesa debido al material de almacenamiento y más rígido de lo normal. Los pulmones son como un globo que nunca se ha hinchado antes por lo que se necesitará un mayor esfuerzo para conseguir que se expanda.
Hay un aumento de secreciones que son difíciles de expectorar ya que la restricción en los pulmones dificulta la toma del aire necesario para toser de forma adecuada. Cuando los pulmones no se vacían de forma correcta por la expectoración, hay un mayor riesgo de infección que puede llevar a la existencia de lesiones en las vías respiratorias aumentando la obstrucción.
Muchos niños con MPS I respiran muy ruidosamente incluso cuando no hay infección. Por la noche pueden estar intranquilos y roncar. A veces un niño puede dejar de respirar durante períodos cortos mientras duerme (apnea del sueño). Esta respiración ruidosa que se detiene y vuelve a empezar puede desarrollar problemas en una de las válvulas del corazón pero es posible que tengan una válvula del corazón afectada durante años sin notar ningún efecto negativo. Si la condición empeora se puede reemplazar las válvulas dañadas con una intervención.
El “soplo cardíaco” puede asustar a los padres pero muchos niños pueden respirar así durante años.
Las dolencias cardíacas son bastante comunes en los individuos con Hurler clásico pero es posible que no se desarrollen o causen los problemas hasta más tarde. Su médico les puede recetar medicamentos para ayudar a aliviar la condición. Las válvulas del corazón están diseñadas para cerrarse herméticamente mientras la sangre pasa de una cámara del corazón a la otra para que no refluya en la dirección equivocada. Si una válvula está debilitada o ha modificado su forma, no puede cerrarse de forma hermética y una pequeña cantidad de sangre puede escapar.
En pacientes muy afectados los músculos del corazón también sufren daños por el depósito de mucopolisacáridos (cardiomiopatía) y el corazón puede sufrir estrés por la obstrucción de las vías respiratorias superiores y las repetidas infecciones pulmonares, o al tener que bombear la sangre a través de unos pulmones más rígidos de lo normal. Algunos niños afectados tienen hipertensión arterial.
El hígado y el bazo en niños con MPS I son más grandes de lo normal debido al depósito de mucopolisacáridos. El hígado grande no causa problemas ni lleva a una insuficiencia hepática pero su volumen puede interferir con el abdomen debido a la postura del niño, la debilidad de los músculos y el mayor tamaño del hígado y bazo.
Hay la posibilidad de que parte de la materia abdominal presione hacia fuera y provoque un pequeño bulto en la pared del abdomen. A esto se le llama hernia que en ocasiones precisa de intervención quirúrgica.
Los individuos con la enfermedad de Hurler tienden a tener la piel gruesa y dura sin elasticidad. Suelen sudar más y tienen las manos y pies fríos debido a que con el paso del tiempo la parte del cerebro que controla la temperatura queda afectada.
A nivel de la columna vertebral las vértebras en la enfermedad de Hurler y Hurler Scheie las vértebras no están correctamente formadas. A veces una o dos vértebras de la mitad de la espalda son más pequeñas, están mal formadas y pueden estar desplazadas hacia atrás. Este desplazamiento hacia atrás de las vértebras puede causar una curva angular (giba), que se detecta en las radiografías. Este problema ocasionalmente necesita tratamiento.
Algunos pacientes con Scheie o Hurler Scheie sienten dolor en la parte baja de la espalda. Esto parece ser debido a que las vértebras inferiores se aplastan y comprimen las raíces nerviosas lumbares. Esto puede diagnosticarse fácilmente mediante una radiografía.
De vez en cuando algunos niños con Hurler sienten dolor en la parte posterior del cuello. Esto puede aliviarse frotando con lociones adecuadas y a muchos niños les gusta que les den un masaje suave en el cuello. Muy raramente los huesos de la parte superior del cuello son inestables. Los Rayos X pueden descubrir si este problema está presente o si va a desarrollarse.
La rigidez en las articulaciones es común en todas las formas de la enfermedad. Todas las articulaciones se ponen rígidas y su movimiento puede quedar limitado. Más tarde en la vida del individuo la rigidez de articulaciones puede causar dolor que se soluciona con la aplicación de calor moderado y analgésicos normales. El movimiento limitado en los hombros y brazos puede dificultar la acción de vestirse. Los individuos con MPS I pueden tenerlas caderas luxadas.
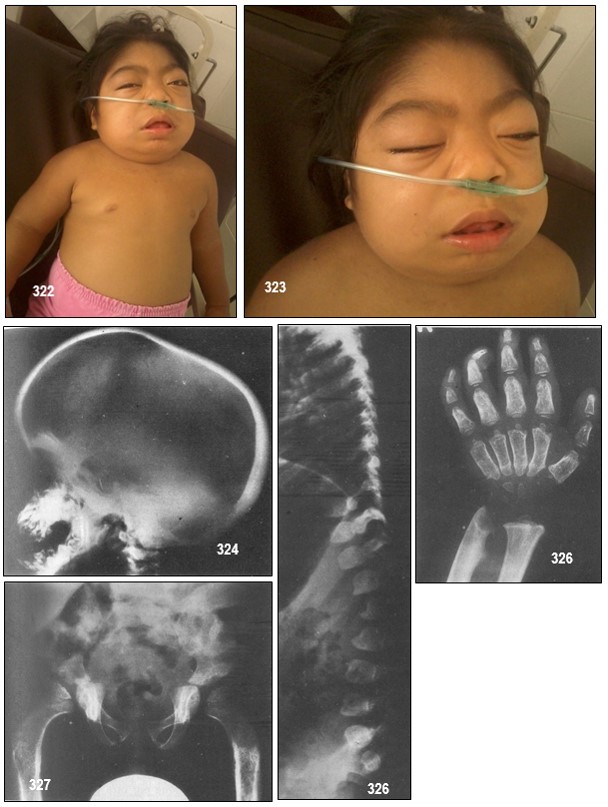
La forma de la mano es muy característica y se ha usado para el logotipo de la Asociación MPS. Las manos son anchas con dedos gordos que gradualmente se van curvando pareciendo garras; véase la radiografía de la mano del caso clínico que presento.
Los individuos con Scheie a veces sienten dolor y pierden sensibilidad en las yemas de los dedos; a esto se le llama Síndrome del Túnel Carpiano. La muñeca o el carpo tienen ocho huesos pequeños conocidos como huesos carpianos, que están unidos por tiras de proteína fibrosa llamadas ligamentos.
Los nervios tienen que atravesar la muñeca entre los huesos carpianos y los ligamentos. Si los ligamentos se agrandan por acúmulos de mucopolisacáridos los nervios quedan comprimidos y esto puede causar un daño irreversible del nervio.
Un nervio dañado hará que el músculo en la base del dedo pulgar se atrofie y pierda fuerza muscular.
Se sabe ahora que los/as niños/as con Hurler y Hurler Scheie también pueden tener este problema. Si su hijo/a parece tener el dolor en las manos, particularmente por la noche, es recomendable que se haga una prueba eléctrica llamada “estudio de conducción de nervio” que mostrará si el Síndrome del Túnel Carpiano es la causa. Se puede operar para liberar el nervio y recuperar la fuerza muscular.
Pruebas y exámenes.- ECG. Pruebas genéticas para el gen de la alfa-L-iduronidasa (IDUA). Exámenes de orina para determinar mucopolisacáridos adicionales. Radiografía de la columna.
Tratamiento.– La terapia de reemplazo enzimático le agrega una forma funcional de la enzima faltante. Se ha utilizado un trasplante de médula ósea en algunos pacientes con esta afección y el tratamiento ha tenido resultados variados. Otros tratamientos dependen de los órganos que están afectados.
El nombre genérico del producto que se utiliza como la Terapia Enzimática Sustitutiva en la MPS es Aldurazyme ® y realizado por el laboratorio Genzyme a Sanofy Company. Este producto ha llevado a cabo amplias pruebas clínicas incluyendo pruebas y estudios con control de placebo en las que se proporcionaba infusiones intravenosas a grupos de pacientes. Parte de estos estudios se llevaron a cabo en el Reino Unido y la mayoría de los pacientes que se incluyeron tenían la variante atenuada de MPS I o Hurler-Scheie.
Aunque es muy temprano para conclusiones definitivas, hasta ahora los resultados de estudios clínicos con Aldurazyme ®han sido muy positivos mostrando una reducción de los problemas ortopédicos asociados al síndrome de Hurler tales como una mayor movilidad de las extremidades y una mejoría en la rigidez de las articulaciones. También se han visto mejorías significantes en la función respiratoria, uno de los síntomas más típicos en todas las variantes de MPS I. Aún hay cuestiones por resolver sobre la eficacia de la Terapia Enzimática Sustitutiva.
La enzima no puede atravesar la barrera hematoencefálica y llegar al cerebro por ello cualquier mejoría experimentada en niños y niñas con MPS I tipo severo utilizando la T.E.S. están limitados a mejorar su calidad de vida mediante un alivio de los síntomas no neurológicos.
Es posible también utilizar la Terapia Enzimática Sustitutiva juntamente con el Trasplante de Médula Ósea para mejorar la condición física general del niño antes del trasplante. Se administra la TRE hasta poder realizar el Trasplante de Médula Ósea y después durante unos meses, hasta observar que el trasplante funciona.
Pronóstico.- El síndrome de Hurler es una enfermedad con un pronóstico de alentador. Los niños con esta enfermedad presentan problemas del sistema nervioso y pueden morir a temprana edad. Los expertos recomiendan la asesoría genética y la realización de exámenes para parejas con antecedentes familiares del síndrome de Hurler que estén pensando en tener hijos.
MUCOPOLISACARIDOSIS TIPO IV O ENFERMEDAD DE MORQUIO.
INFORMACIÓN BÁSICA.- Mucopolisacaridosis Tipo IV. Síndrome de Morquio (OMIM 25300).- El síndrome Morquio es un desorden de almacenamiento también conocido como Mucopolisacaridosis de tipo IV (MPS IV), también conocida como enfermedad de Morquio en recuerdo del pediatra uruguayo Luis Morquio, de Montevideo, Uruguay, quien en 1929 describió una familia con cuatro niños afectados por la misma condición y que la describió por primera vez, es una enfermedad congénita causada por la deficiencia de la enzima N-acetilgalactosamina 6 sulfatasa (MPS IV tipo A) o de la enzima B-Galactosidasa (MPS IV tipo B). Estas anomalías enzimáticas tienen como consecuencia que se acumulen en diferentes tejidos del organismo cantidades elevadas de mucopolisacáridos. Se incluye dentro de las tesaurismosis o enfermedades por depósito lisosomal. Su frecuencia es de 1:100.000 nacimientos aproximadamente y está considerada una enfermedad rara.
Produce anomalías esqueléticas graves (como veremos más adelante), pérdida de la audición, anomalías visuales por opacidad de la córnea, lesiones hepáticas, cardiacas y respiratorias. Por todo ello la esperanza media de vida de las personas afectadas es de 40 años.
Esta misma condición fue descrita ese mismo año por el Dr. Brailsford en Birmingham, Inglaterra, por lo que es conocido a veces como Síndrome Morquio-Brailsford. El Síndrome Morquio se caracteriza por estatura corta, enfermedad severa en los huesos y falta de preservación de inteligencia.
La deficiencia en dos enzimas puede causar MPS IV, cada una de las cuales tienen un espectro grande de severidad clínica desde leve hasta severa.
Mucopolisacaridosis
Por otro lado, un niño diagnosticado recientemente es importante que se recuerde que el Síndrome Morquio varía mucho en el tipo de severidad de la enfermedad; niños de la misma familia pueden ser afectados diferentemente. El rango de problemas posibles, pero esto no significa que niño va a tener todos los síntomas. Algunas complicaciones pueden salir a muy temprana edad, pueden ocurrir después o simplemente nunca aparecer.
Hasta hoy en día, no existe ninguna cura para individuos afectados de estos desórdenes, pero existen maneras de manejar los diferentes retos que tendrán y ayudarles a disfrutar de la vida. El transplante de médula ósea ha sido intentado para el tratamiento de MPS IV, pero con muy poco éxito. La comunidad científica continúa buscando una forma mejor y más efectiva para tratar estos desórdenes para darles más oportunidades a estos individuos en el futuro.
Las dos variedades de la enfermedad A y B presentan síntomas similares y solo se distinguen en estudios de laboratorio que demuestren cual es la enzima ausente.
Sinónimos:
- -Síndrome o enfermedad de Brailsford.
- -Síndrome o enfermedad de Morquio.
- -Síndrome de Morquio-Brailsford
- -Síndrome de Morquio-Ullrich
- -Mucopolisacaridosis tipo IVA
- -Deficiencia de galactosamina-6-sulfatasa
- -Mucopolisacaridosis tipo IVB
- -Deficiencia de beta galactosidasa
- -Condrodistrofía atípica
- -Condrodistrofia congénita tarda.
- -Condrodistrofia tarda
- -Condroosteodistrofía.
- -Disostosis encondral metaepifisaria.
- -Distrofia ósea familiar.
- -Condrodisplasia hereditaria.
- -Osteocondrodistrofia hereditaria.
- -Disostosis encondral politópica hereditaria
- -Enfermedad de almacenamiento de mucopolisacáridos IV
- -Osteochondrodystrophia deformans.
- -Displasia espondiloepifiseal.
- -Osteocondrodistrofia
Al igual que el síndrome de Sanfilippo, los síntomas pueden aparecer al año de vida, con una expectativa de vida que puede exceder los 20 años, llegando incluso a los 50-60 años en las formas benignas. A diferencia del síndrome de Sanfilippo, el desarrollo mental tiende a ser normal. Tiene una muy baja incidencia, y en nuestro país desconocemos su frecuencia.
Causas: Los mucopolisacáridos están compuestos por una cadena larga de moléculas de azúcar utilizados para la formación de los huesos, cartílagos, piel, tendones y muchos otros tejidos en el cuerpo. Estos forman parte de la estructura en el cuerpo y también proveen de esas características especiales que hacen que el cuerpo funcione adecuadamente.
Por ejemplo, el líquido viscoso y resbaloso que lubrica las articulaciones es en parte hecho de mucopolisacáridos. El cartílago huloso en las articulaciones es otro ejemplo. Todos los tejidos tienen en algún grado algo de esta sustancia como parte normal de su estructura.
Una palabra más moderna para mucopolisacáridos es glicosaminoglicanos o GAG, que significa glucoaminoglucoso polímero o cadenas repetitivas de azúcar encontradas en estos materiales. Estas cadenas de azúcar son submicroscópicas y no pueden ser vistos con el ojo, pero pueden ser estudiados con instrumentos científicos y métodos analíticos.
Para entender cómo es que la acumulación de GAG puede causar MPS IV, es importante entender que en el curso de una vida normal existe un proceso continuo de formación de nuevos mucopolisacáridos y dispensar de los viejos – es un proceso de reciclaje.
Este proceso continuo es necesario para mantener la salud. Los procesos de descomposición químicos y reciclaje requieren una seria de herramientas bioquímicas llamadas enzimas. Para poder descomponer GAG, se necesitan una seria de enzimas o herramientas trabajando en secuencia una detrás de la otra.
La cadena de GAG se rompe al remover una molécula de azúcar mientras otra se forma al mismo tiempo al final de la cadena. Cada enzima tiene en este proceso un propósito específico en el cuerpo con una acción muy especial, de la misma forma como un atornillador trabaja con tornillos o un martillo con clavos. Individuos que tienen MPS IV son personas que no tienen alguna de las dos enzimas específicas, las cuales son esenciales para la descomposición de uno de los GAG llamados sulfato keratan.
El sulfato keratan que no ha podido descomponerse totalmente se queda almacenado dentro de las células en el cuerpo y comienza a aumentar causando daño progresivo. El GAG por sí mismo no es tóxico pero la cantidad de éste y el efecto de almacenarlo por tanto tiempo dentro del cuerpo llevan a los problemas físicos recurrentes.
Patológicamente la cantidad anormal de sulfato de keratán es excretada en la orina, disminuyendo sus niveles a medida que aumentan en edad. Los pacientes con este síndrome aparentan ser normales al nacimiento.
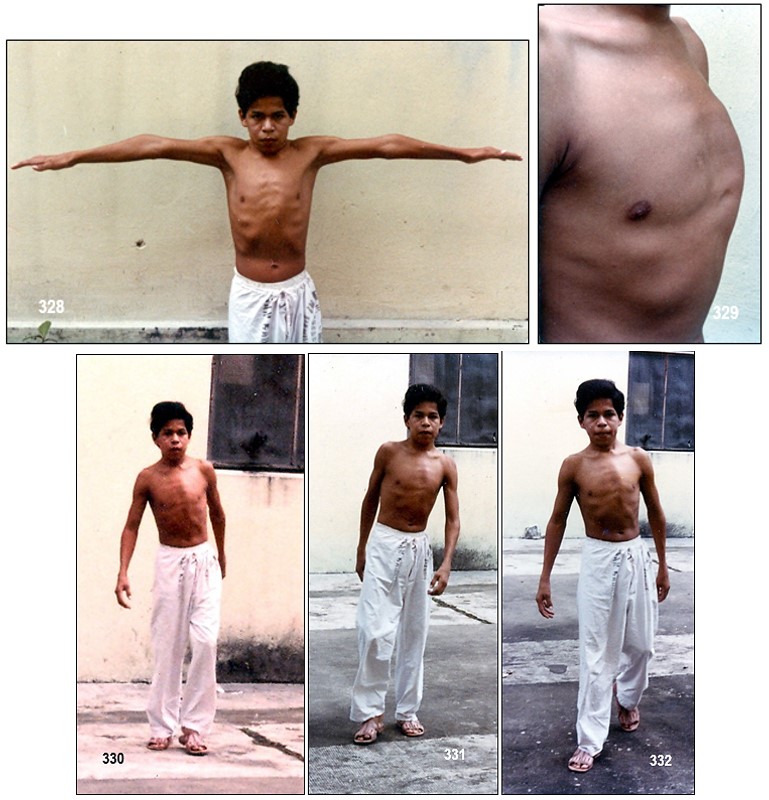
Las anomalías típicas esqueléticas incluyen: enanismo con tronco corto, tórax en carinatum, cifosis, hiperlordosis, escoliosis, deformidad ovoide de las vértebras, genu valgus, deformidad epifisaria de los huesos largos, metáfisis ancha y osteoporosis. La hipoplasia odontoides es un hallazgo clínico universal, la inestabilidad de la apófisis odontoides puede resultar en una subluxación atlantoaxial y compresión de la médula espinal con consecuencias médicas graves. Entre las manifestaciones extraesqueléticas se citan: opacidad corneal, hepatomegalia, lesiones valvulares cardíacas; la apariencia facial es característica con prognatismo, nariz corta y dientes espaciados con defectos en el esmalte, y macroglosia; la pérdida de la audición ha sido reportada como frecuente.
En vista de la poca incidencia de este síndrome, reportado en la literatura médica a nivel mundial y a las escasas publicaciones en cuanto al manejo anestésico, hemos querido presentar este síndrome a propósito de un caso, manejado en nuestra institución, donde en su historia no se habían hecho reportes de un manejo anestésico previo de esta enfermedad, donde la técnica anestésica endovenosa fue la planificada, previendo las patologías agregadas de dicha alteración metabólica.
Genética: Existen dos diferentes tipos de deficiencias enzímicas que se han encontrado como causa del síndrome de Morquio. Cada uno de los desórdenes se llama tipo A y tipo B. Los nombres de las enzimas son N acetylgalactosomina 6 sulfatado (tipo A), también llamado galactoso 6´sulfatado, y beta galactosidasa (tipo B). MPS IV A, que es la forma más común, fue primeramente reconocido como la forma severa del desorden. MPS IV tipo B fue inicialmente considerado como una forma “atenuada” (se usa este término en vez de “leve”, ya que los afectos de estas patologías la enfermedad es bastante significativa para que sean considerados “leves”) una variante del síndrome del Morquio.
Ahora se ha reconocido que la forma severa y atenuada del síndrome no son causados por la deficiencia de una misma enzima, pero que cada uno de estos tipos tienen en sí mismos su propio espectro de severidad clínica. Esto ocurre como resultado de las mutaciones de pérdida de función en ambas copias del gen GALNS. Se han detectado diferentes mutaciones en GALNS y existe cierta correlación genotípica-fenotípica – lo que significa que, en cierta forma, las mutaciones presentes predicen la severidad de las manifestaciones clínica. Existe una gran variabilidad de expresión clínica en ambos subtipos.
En general, individuos que tienen MPS IV tipo B tienen problemas similares, pero tienden a ser afectados más levemente que los que tienen MPS IV tipo A. Esta variante obedece a una mutación en el gen W273L GLB1. Ambas enzimas involucran la descomposición química del sulfato keratan, que es un tipo de glicosaminoglicano. El sulfato keratan es primariamente encontrado en los huesos y los tejidos. La acumulación del sulfato keratan en los huesos y tejidos es responsable por la mayoría de los problemas que afectan a los pacientes con ambos tipos de MPS IV.
Frecuencia: El síndrome del Morquio es uno de los desórdenes de mucopolisacáridos más raros en los Estados Unidos. Figuras confiables de incidencia de esta enfermedad no se encuentran disponibles, pero los estimados varían entre 1 de cada 200,000 nacimientos vivos a 1 de cada 300,0000 nacimientos vivos. Aun cuando estos desórdenes son raros, cada paciente requiere de tanto cuidado intensivo que el impacto de esta enfermedad es alto en el sistema médico.
La creencia común es que una enfermedad genética es un problema de salud que se pasa de padre o madre a sus hijos por generaciones. Mientras sí es cierto que muchas enfermedades genéticas se pasan por generaciones, algunas enfermedades están ocultas o recesivas y se manifiestan cuando los dos genes de un individuo son afectados. MPS IV es una enfermedad de este tipo. La mayoría de las familias que tienen un niño con MPS IV no tienen en su historia familiar ningún problema genético – el MPS IV aparece de pronto.
Para entender mejor esta enfermedad, es importante aprender un poco sobre genética. Todos los humanos estamos formados con dos grupos de genes completos, uno de la madre y otro del padre. De esta forma, un individuo siempre tendrá la mitad de sus genes de la madre y la otra mitad de su padre.
Estos grupos de genes juntos forman un 100% de los genes requeridos para vivir. Por cada enzima creada en el organismo hay dos genes, uno de la madre y otro del padre. Para la mayoría de las enzimas, si uno de los genes trabaja bien, esto es más que suficiente para mantener a este individuo saludable. Básicamente, la enzima puede hacer el doble del trabajo con una mitad.
Sin embargo, si ninguno de los genes para una enzima en particular trabaja adecuadamente, éste individuo tendrá muy poco o nada de esta enzima en su cuerpo y sufrirá a través de su vida la carencia de esa enzima.
Debido a que los padres de un niño que tiene MPS IV cada uno tiene otro gene que sí trabaja, las posibilidades son 3 de 4 embarazos resultarán con un niño que tiene al menos un gene normal y no enfermedad. También tienen 1 de 4 posibilidades que todos los embarazos resulten con un niño que tiene un gene defectuoso de cada uno de los padres y sean afectados por la enfermedad. Por otro lado, los hermanos y hermanas saludables de un niño con MPS IV tienen 2 de 3 posibilidades de que sean portadores. Los portadores de esta enfermedad tienen un gene bueno y otro defectuoso.
En general, esta enfermedad es tan rara que las posibilidades de que un portador se case con otro portador son muy bajas.
Síntomas.- La enfermedad de Morquio tiene un amplio espectro de manifestaciones clínicas. La expectativa de vida depende de la gravedad clínica, lo que se correlaciona con la actividad enzimática residual en los casos más atenuados, o nula en los más serios. La mortalidad se relaciona directamente con la inestabilidad atlanto-axoidea, la mielopatía cervical secundaria y las enfermedades respiratorias (apnea del sueño, hipertensión pulmonar e infecciones recurrentes). Se han reportado casos de supervivencia hasta los 70 años; sin embargo, la expectativa general se encuentra entre la tercera y la cuarta décadas de la vida.
Además, la clínica depende del tipo de mucopolisacárido acumulado, pero generalmente tienen implicaciones similares, caracterizándose por un curso degenerativo letal progresivo con presencia de deformidades óseas, retardo del crecimiento, retraso madurativo y mental, sordera y alteraciones oculares, entre otras alteraciones.
A continuación se resumen las manifestaciones clínicas:
- -Rasgos faciales toscos (más leves que en el síndrome de Hurler o el de Hunter)
- -Macrocefalia
- -Dientes ampliamente espaciados (diastemas). Con alteración del esmalte
- -Tórax en forma de campana con las costillas ensanchadas en la parte inferior
- -Baja estatura con un tronco especialmente corto
- -Articulaciones hipermóviles
- -Desarrollo anormal de muchos huesos, incluyendo la columna vertebral, pudiendo producir compresión de la médula espinal (puede conducir a debilidad o parálisis) y manifestaciones neurológicas periféricas.
- -Opacidad corneal
- -Cardiopatías
- -Problemas respiratorios
Diagnóstico prenatal: Son enfermedades que ya están presentes desde el nacimiento, y suelen aparece tempranamente, generalmente ya son evidentes al nacimiento o durante el primer o segundo año de vida; aunque pueden no hacerse evidentes hasta etapas más tardías.
Si existe el antecedente familiar de un niño con MPS IV, existe la posibilidad de hacer exámenes durante los embarazos subsecuentes para saber si el feto está afectado por la enfermedad. Por eso es muy importante las valoraciones y controles etapas tempranas del embarazo. Las muestras del amniocenthesis y charionic villus pueden ser hechos para diagnosticas MPS IV en el útero.
Diagnóstico: El diagnóstico de la enfermedad de Morquio puede hacerse por las características físicas si estas son evidentes, mediante determinación de la sustancia acumulada en los tejidos o líquidos corporales (orina), detección del déficit enzimático en muestras de sangre o celulares (leucocitos y fibroblastos) dependiendo de cada caso, por estudios genéticos, e incluso de forma prenatal con estudios del líquido amniótico. Estas enfermedades no tienen un tratamiento eficaz, y pronóstico fatal, por lo que es importante la prevención, y en este apartado, la mejor arma del que disponemos es el consejo genético. Es por eso que los estudios genéticos, más que diagnósticos de la enfermedad, nos servirán para detectar a los posibles portadores dentro de la familia de un afectado.
Hallazgos radiológicos: Los hallazgos radiológicos varían con la edad del paciente; es la única mucopolisacaridosis que no presenta cambios óseos similares a la disostosis múltiple. Los cambios más representativos son la platiespondilia, la hipoplasia odontoidea, la coxa valga y el aplanamiento progresivo de la cabeza femoral; los metacarpos son cortos y anchos.
Una complicación peligrosa, secundaria a la deformidad ósea, es la compresión medular, seguida de subluxación atlantoaxoidea; los signos neurológicos de compresión pueden incluir debilidad, dificultad en la marcha, paraplejia, pérdida de la sensación vibratoria de las extremidades e hiperreflexia.
Problemas clínicos del MPS IV
-Crecimiento: Niños afectados con el síndrome del Morquio usualmente crecen normalmente al principio, pero su crecimiento comienza a retrasarse cerca de los 18 meses de edad. Aquellos individuos afectados severamente por esta enfermedad paran de crecer totalmente cerca de los 8 años de edad. La altura máxima es más o menos entre 90,5 a 1.20 mts.
Otros individuos con Morquio continúan creciendo hasta su adolescencia y pueden alcanzar 1,50 mts. de altura. El tronco del cuerpo es relativamente más corto que las extremidades.
- –Inteligencia: Los individuos que tienen Morquio tienen una inteligencia normal.
- -Apariencia Física: La cara es alterada a cierto punto debido a la enfermedad. La boca tiene a ser más ancha, la quijada es cuadrada y el puente de la nariz es plano. El cuello es muy corto, pero la textura de su cabello no es afectado como en otros desórdenes de MPS.
- -Nariz, garganta, pecho y problemas de oído: Si se mira a un diagrama del esqueleto se verá que el esternón está unido con la espina dorsal por medio de las costillas. En el síndrome del Morquio, el crecimiento de la espina dorsal es afectada.
El esternón continua creciendo normalmente, pero debido a que está pegado a la espina, es forzado a doblarse hacia afuera en una curvatura redonda o algunas veces en forma de pico. El pecho es con forma de campana y las costillas que están fijas en una posición horizontal, causan cierta restricción para respirar eficientemente. Un examen de funcionalidad de los pulmones puede ser hecho para determinar cuanta restricción esta anormalidad en los huesos está afectando la respiración.
Aun cuando los niños con síndrome de Morquio no son necesariamente más propensos a infecciones respiratorias, la restricción en el pecho significa que son menos capaces de confrontar una infección de pulmón. Es importante prestar atención si es que el niño está comenzando con síntomas de infección de vías respiratorias para la prescripción de un antibiótico en caso necesario. Se han reportado casos de pacientes de Morquio que han sufrido problemas severos de respiración después de pasar algunos días en altitudes altas. Es siempre aconsejable consultar si está planeando tomar viajes largos por avión o una vacación en las montañas.
-Tratamiento de enfermedades respiratorias: Los fármacos afectan diferentemente a los pacientes que tienen MPS IV, por lo tanto es esencial que no use medicamentos sin prescripción médica. Medicamentos para controlar la producción mucosa pueden ser no efectivos. Medicamentos, tales como antihistamínicos, pueden secar la mucosa y hacerla espesa lo que provoca que sea más difícil de extraer. Los descongestionantes usualmente contienen estimulantes que pueden elevar la presión sanguínea y estrechar los vasos sanguíneos, ambos efectos son muy indeseables para pacientes con MPS IV. Los antitusígenos que tienen sedantes pueden causar más problemas con apnea al deprimir el tono del músculo y la respiración.
Aun cuando la mayoría de los pacientes que tienen resfríos no requieren de antibióticos, los individuos con MPS IV casi siempre terminan teniendo infecciones bacteriales secundarias causando sinusitis o infecciones del oído medio. Estas infecciones deben ser tratadas con antibióticos. Si los senos y el oído medio no han sido drenados esto hace más difícil superar las infecciones y por lo tanto las infecciones mejoran con los antibióticos, pero luego reaparecen después de que el antibiótico deja de ser efectivo. Algunas veces una terapia de antibióticos crónica puede ser utilizada para ayudar a aquellos individuos que tienen infecciones de oído recurrentes.
Tubos de ventilación también pueden ser usados para mejorar el drenaje de los oídos y acelerar la resolución de las infecciones. Es muy importante consultar un Otorrinolaringólogo que tenga experiencia en individuos de MPS IV para determinar si los tubos son lo mejor.
Muchos pacientes con MPS IV pueden convertirse en alérgicos a los antibióticos o empezar a tener resistencia las infecciones. En estos casos suele recetarse otros antibióticos para ayudar a manejar este problema. Mientras que es cierto que el abuso de antibióticos no es aconsejable, individuos con MPS IV requieren de este tratamiento para la mayoría de las infecciones.
–Boca: Los pacientes con MPS IV tienen una barbilla prominente, boca ancha y su lengua es alargada. Los dientes pueden ser esparcidos (diastemas) con esmalte muy pobre.
Es muy importante que los dientes sean cuidados adecuadamente, debido a la caída de los dientes puede ser muy dolorosa. Los dientes tienen que ser limpiados regularmente. Limpiar dentro de la boca con una esponja en un palillo empapado con un enjuague bucal, ayudará a mantener la boca fresca y evitará el mal aliento. Aun con el mejor tratamiento dental, abscesos se pueden desarrollar alrededor de un diente debido a la formación anormal de los dientes.
En algunas ocasiones las únicas señales de que existe un problema con un diente infectado es cuando el individuo está muy irritado o llorando. Si un individuo con MPS IV tiene una miocardiopatía, es aconsejable que se le den antibióticos antes y después de un tratamiento dental. Esto es debido a que ciertas bacterias en la boca pueden ingresar en la sangre y causar una endocarditis infecciosa. Si dientes tienen que ser removidos con anestésico, es mejor hacerlo en un hospital bajo el cuidado de un anestesista con experiencia y un odontólogo.
–Corazón: Cardiomiopatías pueden ocurrir en paciente con Morquio. Una manifestación constante pero tardía es la regurgitación aórtica, la cual asociada a la seria escoliosis que genera problemas respiratorios, constituyen las principales causas de mortalidad de los afectados. La regurgitación genera un soplo si la válvula, en este caso, la aorta, ha sido dañada por la concentración de mucopolisacáridos.
Muchos pacientes con síndrome de Morquio desarrollan problemas de bloqueo cardiaco. Algunos pueden desarrollar problemas en la válvula aortica. Estos pueden progresar lentamente por años sin ningún efecto clínico aparente. Si esta condición empeora, una operación puede ser necesaria para reemplazar las válvulas dañadas.
Debido a que problemas del corazón ocurren que ocurren en la MPS IV, es necesario hacer un ecocardiograma doppler anualmente (o con la frecuencia que el médico indique), para determinar si algún problema se está desarrollando.
–Abdomen y hernias: En pacientes con MPS IV, el abdomen se “sale” debido a la postura, los músculos abdominales débiles y la hepatoesplenomegalia contribuyen a este abultamiento abdominal. Con frecuencia el contenido del estómago se empuja detrás de un punto débil en la pared abdominal ocasionando hernias. Las hernias pueden ser umbilicales o inguinales. Las hernias de la ingle deben ser reparadas con una operación y pueden recurrir. Las hernias umbilicales no son tratadas usualmente a menos que sean pequeñas y causan el cierre del intestino o son muy grandes provocando problemas más serios. Es común tener recurrencia de una hernia umbilical después de que ésta ha sido tratada, a esta situación se la ha llamado “diátesis herniaria”.
-Huesos y articulaciones: Pacientes con MPS IV tienden a tener problemas significativos en el crecimiento y la formación de hueso. El problema del hueso en MPS IV es diferente al visto en otros desórdenes de MPS. Las características clínicas de MPS IV están relacionadas a los huesos y su efecto en el sistema nervioso si los nervios están comprimidos por el movimiento anormal de los huesos.
–Columna vertebral: Las vértebras normalmente están alineados con el cuello y los glúteos. En individuos con MPS IV. La columna vertebral tiende a ser severamente afectada y las vértebras son anormalmente aplanadas. Generalmente las vértebras son muy malformadas y no interactúan bien entre ellas. Si esto ocurre, las vértebras pueden comprimir la espina dorsal. Una o dos de las vértebras en la parte baja son algunas veces más pequeñas que el resto y afectar la alineación hacia atrás. Esta alineación hacia atrás puede causar que se desarrolle una curva angular, pero no siempre necesita tratamiento quirúrgico. Si esta curvatura no es muy severa, se puede corregir con un braguero puede ser recomendado. Hay diferentes opiniones en cuanto si una operación es necesaria o un braguero es más adecuado para corregir el problema de una columna vertebral curvada.
Si un braguero es utilizado debe ser del tipo que no restringa el movimiento del pecho.
–Cuello: Problemas en esta área son los más serios que personas con este síndrome confrontan. El problema del cuello debe ser discutido inmediatamente que éste ha sido diagnosticado, debido a que estos pueden ser de mucha relevancia una vez que la persona cumpla 5 o 6 años de edad. Las dificultades empiezan a acrecentarse con el defecto estructural en las vértebras superiores de la columna vertebral, que puede empeorar por los ligamentos sueltos. Los huesos que son los estabilizan la conexión entre la cabeza y el cuello generalmente son malformados en la pacientes con MPS IV.
El adontoide se pega entre la primera y segunda vértebra que es la que da apoyo a la cabeza para moverla para adelante y para atrás. Con la displasia de adontoide, el cuello es sumamente inestable, lo que pone a la espina dorsal en riesgo de vida o muerte.
El cordón dorsal es un grupo de nervios que lleva mensajes al cerebro y al resto del cuerpo. Si el cordón dorsal está comprimido o apretado, los efectos se van empeorando gradualmente en el niño con parálisis o muerte, si no se le da tratamiento.
Los niños con síndrome de Morquio deben ser referidos a un cirujano ortopedista a una edad temprana para monitorear la condición de la espina dorsal. Estudios radiológicos serán hechos con la cabeza doblada para adelante con el cuello recto (vista de extensión y flexión) y será repetido año tras año para monitorearlo. Un estudio de la base del cuello es recomendado en el momento que se diagnostique. Si hay dolor o dolor asociado con debilidad en la parte baja de las piernas, el niño debe tener un estudio del cuello para evaluar si hay un cambio en las vértebras del cuello.
Padres familia pueden alarmarse al pensar que el niño tiene una debilidad en un lugar tan vital y puede causarse daño con una caída. Es imperativo dar consejo sobre los riesgos. Es muy importante mantener un balance entre evitar los riesgos y dejar que el niño tenga una vida lo más normal posible, sin embargo, actividades con saltos mortales, parase en la cabeza o sumergirse deben ser evitados totalmente.
El problema de la columna vertebral puede ser corregido con una operación llamada fusión. Pequeños pedazos de hueso son tomados de las piernas y puestos en el cuello donde eventualmente crecen y forman un apoyo firme, uniendo las dos vértebras superiores que están en la base del cráneo. Estas operaciones de fusión son también hechas en pacientes con otras condiciones, pero hay problemas especiales con los pacientes de Morquio que deben ser considerados. La mayoría de los cirujanos ortopédicos tienen muy poco o ninguna experiencia con individuos que tienen MPS IV.
Se han encontrado que es esencial inmovilizar el cuello en la posición correcta (reducir) por 4 meses mientras los nuevos huesos crecen y se unen a la base del cráneo. Una manera de conseguir esto exitosamente es a través del método halo.
El anillo hecho de metal halo es pegado al cráneo y tiene barras que van hacia el cuerpo con una enyesadura. La operación usualmente implica quedarse en el hospital no más de una semana. El halo es requerido de 3 a 4 meses para permitir que sane y se fusione con la espina. A pesar de que cuidar a un paciente con halo es mucho trabajo, generalmente los pacientes se ajustan extremadamente bien.
-Escoliosis: La escoliosis puede ocurrir y si es severa puede requerir intervención. En general, la fusión con el hueso es la mejor alternativa y varillas no son toleradas muy bien. En cualquier caso, los huesos suaves que la cirugía y recuperación sea dificultosa. Muchos pacientes necesitan múltiples procedimientos.
-Articulaciones: Rigidez en las articulaciones es común en el desorden de MPS, con el rango de movimiento muy limitado. Sin embargo, en MPS IV, las articulaciones son un poco más movibles de lo normal debido a los ligamentos sueltos.
–Caderas: Es común que las caderas se luxen, pero éste no es un problema común y tratamiento puede ser no necesario. Si hay dolor después, cirugía puede ser considerada.
–Piernas y pies: Las rodillas de la mayoría de los individuos MPS IV crecen anormalmente y desarrollan rodillas dobladas (genu valgum). Es posible que las piernas sean corregidas con una operación pero es mejor esperar hasta que el niño deje de crecer. Cualquier operación que se haga antes en edad muy tempranas ha dado resultados muy poco satisfactorios debido a que las piernas regresan a la su posición torcida. Los tobillos pueden quedar débiles y se doblan para adentro en MPS IV.
Ocasionalmente botas especiales o tablillas pueden ser utilizadas, mas generalmente con zapatos adecuados es más que suficiente. Los huesos que componen los arcos de los pies son mantenidos en posición por ligamentos y tendones. Estos tienen a debilitarse, resultando en pie plano. Los dedos de los pies son deformes.
–Hombros: Los hombros se dislocan con frecuencia para abajo haciendo que el paciente no puede subir sus brazos más arriba de su cabeza. Esto raramente ocasiona incomodidad y es más que todo que es más difícil vestirse o atraer la atención de la profesora en la clase.
–Muñecas de las manos: Las muñecas de las manos son alargadas y curveadas. Se pueden soltar o blandas cuando los pequeños huesos carpales no se desarrollan apropiadamente y los ligamentos se relajan. Esto puede significar una pérdida considerable de fuerza en las manos. Se han realizados intentos para corregir este problema por medio de operaciones quirúrgicas poniendo injertos en las muñecas, pero desafortunadamente, esta técnica no ha funcionado. En algunas ocasiones poner pequeñas tablillas de plástico en las muñecas puede ser de ayuda. Escribir puede ser difícil para algunos individuos y puede ser que les sea más fácil utilizar una máquina de escribir o un procesador de palabras.
El síndrome de túnel carpiano es común en pacientes con MPS, pero no se ha reportado como un problema frecuente en los pacientes con MPS IV. Si apareciera dolor o entumecimiento de las manos, especialmente en las noches, sería aconsejable hacer un estudio de conducción nerviosa. Este examen puede determinar si estos síntomas son por causa del síndrome de túnel carpiano. Si las manos están débiles o la masa muscular ha disminuido en la base del pulgar es necesaria la participación de un neurólogo. La mayoría de los individuos afectados por MPS no tienen los síntomas clásicos del síndrome de túnel carpiano, aún cuando hay daño de nervio.
-Problemas neurológicos: Cerebro, sentidos y nervios: Ojos: La cornea puede nublarse debido a la acumulación de mucopolisacáridos lo cual interrumpe la claridad en la córnea. Estas cataratas en la córnea por lo general son pequeñas. Si existe mucha catarata, esto puede reducir la potencia visual, especialmente en luz baja. Algunos pacientes no pueden tolerar luz brillante debido a que las cataratas causa un relejo disparejo. Utilizar sombreros para sol o anteojos de sol puede ser de ayuda.
–Oídos: En algunos tipos de MPS IV existe un grado de sordera. Puede ser sordera conductiva o de nervio o ambas lo que puede empeorarse debido a las infecciones frecuentes en los oídos. Es importante que los individuos con MPS IV tengan sus oídos chequeados regularmente para que si hay algún problema éste pueda ser tratado a tiempo y también puedan aprender y comunicarlo.
-Sordera Conductiva: El funcionamiento correcto del oído medio depende que la presión del tímpano y la del canal externo del oído sea la misma que la atmósfera alrededor. Esta presión se ecualiza con el conducto de Eustaquio.. Si este tubo está bloqueado, la presión detrás del tímpano baja. Si esta presión negativa persiste, el flujo que cubre el oído medio comienza a acrecentarse y con el tiempo se convierte pegajoso, casi como pegamento. Esto es llamado efusión del oído medio.
–Sordera Neurosensorial: En la mayoría de los casos, la causa de sordera de nervio es debido al daño ocasionado a los pequeños células de vello en el oído interno.
Esto puede también manifestarse con sordera conductiva, por lo que se le llama sordera mixta. Ambas sorderas pueden ser manejadas con aparatos de sordera que se encuentran en el mercado. Por lo general se cree que aparatos para la sordera no son muy utilizados en personas con MPS.
Manejo y tratamiento general
-Dieta: No existe ninguna evidencia científica de que alguna dieta en particular ayude a las personas con MPS IV, en especial para síntomas de diarrea que tienen a aparecer y desaparecer naturalmente. Algunas veces se encuentran que los cambios en la dieta modifican la secreción de moco excesivo, la diarrea y la hiperactividad, disminuyan de intensidad. Se puede reducir la ingestión de leche o productos lácteos y azúcar, así como evitar productos que tengan muchos aditivos y colorantes.
–Boca: Los pacientes con MPS IV
Es importante recordar que no existe ninguna dieta que evite la acumulación de mucopolisacáridos porque éste es creado dentro del cuerpo. En virtud de lo anterior, reducir el consumo de azúcar, no de productos con azúcar, no reducirá la acumulación de GAG.
-Vómito: Un número pequeño de niños con MPS IV tienen episodios severos ocasionalmente de vómito al punto que el niño puede deshidratarse. La causa es desconocida pero si su niño tiene este problema lo mejor es que llame a su doctor.
-Terapia física: El entumecimiento de las articulaciones no es una característica de MPS IV, pero tenerlas flojas puede ser un problema. Terapia física es necesaria para ayudar a los individuos con MPS IV empezar a caminar nuevamente después de tener cirugía de cuello, espina o rodilla. Los individuos con MPS IV deben mantener una vida activa lo más posible para retener la fuerza en los músculos y su salud en general.
-Anestesia: los individuos con MPS IV necesitan mucho cuidado al darles cualquier tipo de anestésico y siempre debe ser dado por un especialista. Si la columna vertebral está inestable los individuos con MPS IV corren el riesgo de doblarse el cuello mientras están inconscientes, por lo que deben precauciones especiales.
•Pubertad y matrimonio: Los adolescentes con síndrome de Morquio tiene un desarrollo normal durante la pubertad, pero las mujeres pueden tener retraso para su período menstrual. El síndrome de Morquio no afecta la fertilidad pero una mujer que sea afectada con el síndrome severamente, es aconsejable que no tenga hijos debido a los riesgos. Todos los hijos nacidos de individuos con MPS IV son automáticamente portadores pero no tendrán la enfermedad a menos que la pareja sea también portador.
Vida con síndrome de Morquio: En el pasado, los pacientes afectados severamente no sobrevivían más allá de los 20 a 30 años de edad, sin embargo, los menos afectados han vivido hasta los 50 o 70 años de edad.
Conforme la comunidad médica hace avances y maneja mejor las condiciones que afectan a estos pacientes, la tasa de mortalidad disminuirá. Por lo tanto, es sensato esperar un niño vaya a tener una esperanza de vida razonable.
Al principio cuando se conoce el diagnóstico es muy difícil y puede ser una experiencia devastadora. Este es un desorden severo, pero no hay razón para no encontrar esperanza y confort. Niños con Morquio pueden tener una vida activa y plena- muchos pueden ir a la escuela regular, pertenecer a clubes y otras actividades-. Adultos con este síndrome han ido a la universidad y hasta pueden manejar automóviles. Algunos niños con MPS IV pueden beneficiarse de tener una educación regular y disfrutar de una vida social.
Tratamiento específico para MPS IV: La teoría detrás del tratamiento del desorden de MPS Se demostró por la Dra. Elizabeth Neufeld que pequeñas cantidades de enzimas lisosomales, aun cuando son intracelulares por naturaleza, pueden ser secretadas de células normales. La secreción de estas enzimas puede ser tomada por las células adyacentes y dirigidas al lisosoma donde funciona normalmente. Es así como se demostró que el defecto bioquímico en la célula deficiente de la enzima lisosomal puede ser corregida al tomar pequeñas cantidades de esta enzima secretada de la célula normal adyacente. Este fenómeno es conocido como “sección cruzada”, y es lo que hoy en día está siendo la base de la estrategia de tratamientos que se están desarrollando para este síndrome.
Terapia de reemplazo enzimático: Una prueba clínica de reemplazo de enzima fue conducido para pacientes con MPS I. Para esta enfermedad, en el 2003 la FDA aprobó el uso de la terapia enzimática sustitutiva con laronidasa, la forma recombinante de la α-L-iduronidasa (Aldurazyme®) obtenida por ingeniería genética, esta enzima cataliza la hidrólisis de los enlaces α-.L-iduronosidos en el dermatan sulfatado, esta enzima ha sido una gran esperanza para aquéllos niños cuyo diagnóstico se hizo ya más tarde y no es posible hacerles un trasplante de médula ósea.
El trasplante de la médula se debe realizar en muy comenzar a prevenir el retraso mental.
El trasplante de la médula se realiza después de la radioterapia de cuerpo entero, y es tratamiento muy eficaz del síndrome de Hurler, excepto la enfermedad del hueso y de ojo. Por otro lado, la terapia de reemplazo enzimático, la enzima retrasa el desarrollo de la enfermedad y mejora los síntomas en muchas áreas pero aún no ha sido posible evitar la regresión neurológica y el retraso mental. Para los otros tipos, desde las MPS II a la IV, actualmente se está investigando intensamente la posibilidad de desarrollar una terapia de reemplazo enzimático, para aportar a las personas afectadas la enzima deficiente, de manera que puedan degradar el keratan sulfato acumulado. Se están divulgando los avances en la producción a escala banco de la enzima recombinante GALNS en E. coli BL 21 empleada en terapia de reemplazo enzimática del síndrome de Morquio.
Por otro lado, están en marcha pruebas terapéuticas clínicas para evaluar el beneficio de estas estrategias para las personas afectadas, en un proyecto de la St. Louis University en EEUU. Para la MPS tipo VI la FDA aprobó en el 2005, Naglazyme®, que es una versión elaborada de la enzima ASB. Naglazyme® ha mejorado sustancialmente la actividad y resistencia física de pacientes con este tipo de MPS, que se traduce con una disminución de los GAGs en la orina. Otros tratamientos dependen del sistema de órganos afectados.
Consejo genético: El Síndrome de Morquio es un proceso autosómico recesivo. Esto significa que una pareja que ha tenido un hijo con esta afección tendrá un riesgo de 25% de tener otro hijo con este problema.
En contraste, la persona afectada tiene muy poco riesgo de tener un hijo con la misma afección.
Esto tiene cierta relevancia ya que la fertilidad en las personas afectadas es normal. Las mujeres normalmente llevan sus embarazos a término, a pesar de que es probable que desarrollen compromiso respiratorio posteriormente durante el embarazo y se ven en la necesidad inevitable de someterse a una cesárea.
Bibliografía
- Hopkin RJ, Grabowsi GA: Lysosomal storage diseases, pp 2315–2319. In: Kasper DL et al (eds) Harrison’s Principles of Internal Medicine, 16th ed. McGraw- Hill, New York, 2005.
- McKusick VA: Mendelian Inheritance in Man, 12th ed. 1998 (OMIM at www.ncbi.nlm.nih/Omim).
- Neufeld EF, Muenzer J: The mucopolysaccharidoses, pp 3421–3452. In: Scriver CR et al (eds) The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, New York, 2001. Passarge E et al: Krankheiten infolge genetischer Defekte im lysosomalen Mucopolysaccarid-Abbau. Dtsch Med Wschr 99: 144–158, 1974.
- Asociación de las Mucopolisacaridosis y Síndromes Relacionados, “MPS España “ C/ Anselm Clavé nº 1. Apartado de Correos 6. 08787 La Pobla de Claramunt (Barcelona)www.mpsesp.org – Email: info @mpsesp.orgTefls.93.804.09.59 – 617.080.198 – 661.710.152