INFORMACIÓN BÁSICA.- Micosis Fungoide (MF) (ORPHA: 2584/OMIM:254400/CIE-10:C84.0).- La MF es un linfoma cutáneo de células T epidermotropo, caracterizado por una proliferación de linfocitos T malignos de pequeño o mediano tamaño con núcleo cerebriforme. Inicialmente se manifiesta en la piel y suele quedarse en ella durante años e incluso décadas, para luego en fases avanzadas, comprometer ganglios linfáticos y órganos internos, pudiendo ocasionar muerte del afectado.
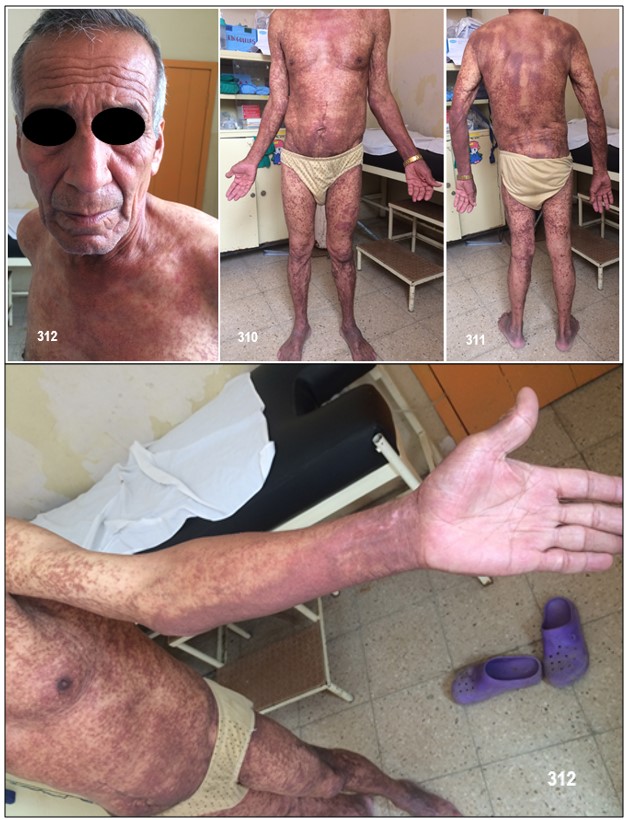
El síndrome de Sézary (SS) es una variante clínica de linfoma cutáneo de células T y está definido por la tríada de eritrodermia, linfadenopatías y presencia de células neoplásicas (células de Sézary) en piel, ganglios linfáticos y sangre periférica.
Clasificaciones.- Hasta fechas próximas los linfomas cutáneos fueron clasificados de acuerdo a las clasificaciones histológicas realizadas por hematólogos y patólogos para los linfomas no Hodgkin ganglionares. En este vía se utilizaron durante muchos años la clasificación de Kiel modificada y la Working-Formulation. La aparición en los últimos años de nuevos tipos de linfomas y la necesidad de utilizar de manera uniforme una misma clasificación por los distintos grupos de trabajo dio lugar a que el Grupo Internacional de Estudio de Linfoma propusiera en el año de 1994 una clasificación nueva denominada Clasificación Revisada Europea-Americana de Neoplasias Linfoides, que posteriormente fue modificada por la OMS en 1999.
No hace mucho tiempo, y apoyándose en la idea de que los linfomas cutáneos primarios deben de ser considerados un grupo distintivo tanto clínico como biológicamente, se publicó la clasificación de linfomas cutáneos primarios por el Grupo de Estudio del Linfoma Cutáneo de la Organización Europea para la Investigación y Tratamiento del Cáncer (EORTC). Esta clasificación se apoyó en una combinación de criterios clínicos, histológicos, inmunohistoquímicos y genéticos, diferenciando las distintas entidades sobre la base de unos criterios que en conjunto permitirían predecir el curso clínico, la respuesta al tratamiento y el obviamente el pronóstico.
Epidemiologia.- La MF representa el tipo más frecuente de linfoma cutáneo primario. Se considera que existe un aumento progresivo en su incidencia, aunque desconocemos si este incremento es debido a un aumento real o, por el contrario, sólo refleja un mejor conocimiento de la entidad y un diagnóstico más precoz. Weinstock y Horm hallaron una incidencia media de 0,3 casos por 100.000 habitantes, representando la MF el 2,2% de todos los linfomas.
La incidencia de la MF se incrementa a medida que avanza la edad y la edad media de presentación es aproximadamente a los 50 años. Hay publicaciones en niños y adolescentes, sin que por ello comporte peor pronóstico. Aparece con mayor frecuencia en varones que en mujeres en una relación de 2,2 varones por una mujer, y la más afectada es la raza negra que la blanca en una proporción de 2 a 1.
Etiología.- El origen de la MF se desconoce. Se han considerado varias teorías, entre las que destacamos las siguientes:
— Virus. Aunque han sido varios los virus implicados en la etiología, como el virus del herpes simple o el de Epstein-Barr, los datos más relevantes relacionaron a la MF con los retrovirus y, en concreto, con el virus linfotropo de células T humanas (HTLV 1 y 2). En la actualidad existe alguna controversia, ya que estudios ocasionales han descartado que el virus HTLV 1 pudiera estar implicado directamente en la etiología o patogénesis del linfoma cutáneo de células T.
— Estimulación antigénica crónica y persistente. Se ha sugerido que la presencia y persistencia de un antígeno, hasta ahora desconocido, causaría una estimulación persistente de los linfocitos T y tras un período variable de tiempo un clon celular proliferaría de manera independiente de los mecanismos reguladores.
— Genética. La ausencia de una agregación familiar bien documentada indica que la MF no es primariamente una enfermedad genética, aunque no se puede descartar cierto grado de predisposición a padecerla. En algunos estudios se ha encontrado cierta asociación con determinados antígenos HLA, principalmente HLA-DR5, y se han descrito anomalías numéricas o estructurales en algunos cromosomas, principalmente en el cromosoma 1, donde además se sospecha que la región entre 1p22 y 1p36 podría contener un gen relacionado con el proceso de transformación y/o progresión de la MF.
Navas y cols. recientemente sugirieron una relación entre hipermetilación del gen p16 (localizado en el brazo corto del cromosoma 9) y curso clínico desfavorable.
Manifestaciones clínicas: Las manifestaciones clínicas de la MF son muy variables. Se pueden agrupar en dos apartados:
- Micosis fungoide clásica (tipo Alibert-Bazin).
- Otras formas clinicopatológicas de micosis fungoide.
- Micosis fungoide clásica (tipo Alibert-Bazin)
La MF clásica o de tipo Alibert-Bazin se divide en tres períodos o fases evolutivas denominadas: premicósica o eritematosa, en placas infiltradas y tumoral. Estas tres fases pueden superponerse entre ellas.
- Fase premicósica o eritematosa
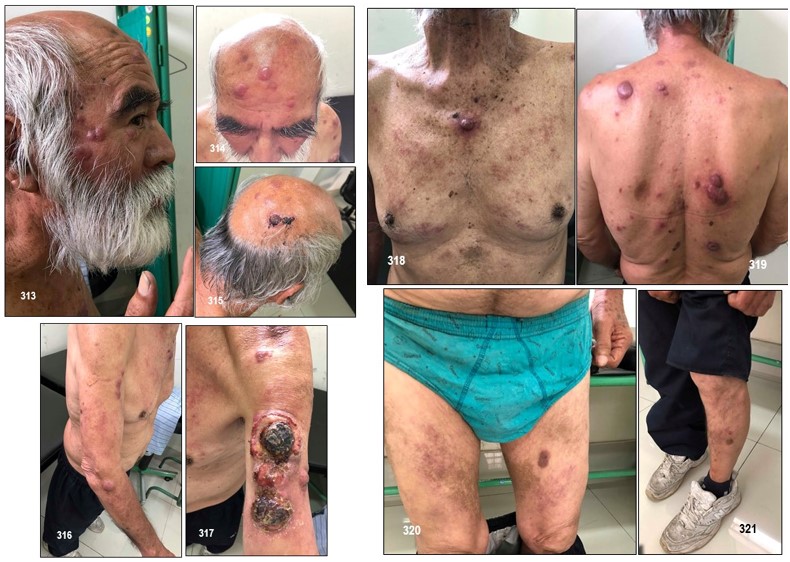
Las lesiones cutáneas son muy variadas. El tipo más común es el de placas de tamaño variable y formas generalmente redondeadas u ovales, eritematosas o eritematoescamosas, a veces con cierto tinte parduzco, poco o nada infiltradas al tacto, estables y de límites bien definidos o difusos e imprecisos (véase la imagen 319). Se localizan con preferencia en tronco y raíz de miembros. En algunos casos las lesiones adoptan otras formas menos frecuentes como manchas con aspecto roseoliforme, placas psoriasiformes o erupciones papulosas e incluso vesiculosas, como un eccema, que hacen de la MF una gran simuladora como en su tiempo se dijo de la sífilis. Un signo de ayuda diagnóstica es la persistencia de islotes reservados de piel sana en el centro de las placas. Todas estas lesiones tan variadas tienen en común el prurito intenso y desagradable, que va provocando secundariamente excoriaciones, eccematización o infecciones secundarias; sin embargo, en algunos casos las lesiones son poco o nada pruriginosas. Esta primera fase puede durar varios años, e incluso décadas, aunque generalmente con el tiempo los elementos se vuelven más numerosos, se extienden y confluyen unos con otros. La duración media desde el inicio de los síntomas hasta el diagnóstico es de unos 6 años.
La entidad conocida como parapsoriasis en grandes placas, caracterizada por placas eritematoescamosas de tamaño grande, a veces con parcelas de atrofia y pigmentación (poiquilodermia atrófica vascular), es considerada por algunos autores como una MF en fase inicial.
2. Fase de placas infiltradas
Esta segunda fase se instaura de forma progresiva con la aparición, sobre lesiones previas o en piel sana, de placas esta vez infiltradas. La infiltración aparece a menudo sobre el borde de ciertas placas, que incluso pueden llegar a formar figuras anulares o serpinginosas, con centro rosado, deprimido, finamente escamoso y borde sobrelevado de color rojo vivo. En otros casos la infiltración es más homogénea, apreciándose una placa bien circunscrita, abollonada y con borde en pendiente suave. La superficie puede ser lisa o ligeramente escamosa, aunque a veces aparece erosionada y con costras.
3.Fase tumoral
La fase tumoral se desarrolla gradualmente y con el tiempo, bien sea sobre piel sana o sobre piel previamente afecta, aparecen tumores que pueden ser hemisféricos o mostrar forma de hongo o seta, por constricción de la base. Son de consistencia elástica o semiblanda. Su superficie es lisa y de coloración rojiza o purpúrica, aunque con frecuencia se ulceran o necrosan, dando lugar a úlceras a veces más o menos profundas.
Los casos descritos como MF tumoral d’emblée (forma tumoral desde el comienzo) se cree actualmente que no son MF tumorales, sino que corresponderían a linfomas T periféricos.
Es muy rara la afectación de mucosas, aunque se han observado placas erosivas e infiltradas, e incluso tumores, en las mucosas bucofaríngea, ocular, digestiva, respiratoria y genital.
-Otras formas clinicopatológicas de micosis fungoide
Además de la MF clásica tipo Alibert-Bazin podemos encontrar otras variantes menos frecuentes como son:
-Micosis fungoide asociada a mucinosis folicular.
La asociación entre MF y mucinosis folicular está ampliamente reconocida en la literatura, pudiendo esta última preceder, concurrir o aparecer posteriormente a la MF.
La clasificación EORTC para los linfomas cutáneos primarios la definió como una variante distintiva de MF caracterizada por infiltrados foliculotropos con degeneración mucinosa del folículo y que afecta preferentemente a cabeza y cuello. La clínica de estos casos es muy polimorfa y se han descrito distintas formas de expresión como alopecia no inflamatoria tipo alopecia areata; pápulas foliculares, que pueden confluir en placas de diferentes tamaños; placas eritematosas e infiltradas, nódulos y tumores, todo ello asociado con frecuencia a pérdida de pelo y prurito.
-MF folicular o quística
En algunos casos el infiltrado linfoide atípico se localiza exclusivamente a nivel peri o intrafolicular, sin epidermotropismo o mucinosis folicular. Este foliculotropismo da lugar a lesiones clínicas diferentes a las observadas en la MF clásica y que consisten en lesiones con apariencia de comedones o quistes miliares, pápulas foliculares, alopecia o placas con aspecto de piel de naranja.
-Reticulosis pagetoide o enfermedad de Woringer-Kolopp.
Se trata de una variante distintiva de MF caracterizada por la presencia de placas, generalmente únicas y localizadas principalmente en miembros, eritematoescamosas, psoriasiformes, con un borde moderadamente elevado y circinado, que a veces muestran un centro más claro o normal. Tiene un curso crónico y su evolución se considera benigna. Existe una forma diseminada (tipo Ketron-Goodman) que tiene una histopatología similar, pero el término de reticulosis pagetoide debe de ser reservado para el tipo localizado o enfermedad de Woringer-Kolopp.
Se ha denunciado, de manera aislada, sobre la progresión del Woringer-Kolopp a la forma diseminada de reticulosis pagetoide, pero esto parece ser algo excepcional.
-MF unilesional
Es una variante de MF caracterizada por lesiones cutáneas solitarias, en forma de placas eritematosas o eritematoescamosas (excepto un caso que presentó un nódulo), de varios centímetros de diámetro, localizadas principalmente en tronco o miembros superiores y de comportamiento generalmente benigno.
El diagnóstico diferencial con la Enfermedad de Woringer-Kolopp puede ser difícil. Para Verret y cols. la enfermedad de Woringer-Kolopp se diferencia de la MF unilesional por la clínica (aspecto más verrucoso, borde policíclico y tendencia a la curación central), histopatología (afectación principalmente epidérmica de linfocitos con citoplasma pálido y voluminoso) e inmunohistoquímica (el fenotipo de los linfocitos es con frecuencia CD8+).
-Micosis fungoide simulando dermatitis pigmentaria y purpúrica
En algunos casos la MF se ha presentado en forma de lesiones purpúricas en miembros inferiores, semejando una dermatitis purpúrica y pigmentaria. Según Lipsker y cols. se debe de sospechar MF en aquellas dermatitis purpúricas y pigmentarias extensas, de más de 1 año de evolución, con patrón reticulado y patch test negativos. Las características histológicas que deben sugerirnos la presencia de MF son:
- — Infiltrado polimorfo, con eosinófilos y células plasmáticas.
- — Poca espongiosis y mayor grado de exocitosis (epidermotropismo).
- — Mayor densidad de infiltrado en banda en dermis papilar.
- — Presencia de linfocitos atípicos, cerebriformes, en el infiltrado.
- -MF eritrodérmica
En algunos casos, bien sobre lesiones previas de MF o de novo sobre piel sana (hombre rojo de Hallopeau), aparece un cuadro de eritrodermia. No existe unanimidad respecto a si la eritrodermia como expresión de MF es o no diferente a la que aparece en el SS.
Otras formas.- En la literatura dermatológica existen descripciones de distintas formas de presentación de la MF poco frecuentes, como, por ejemplo, hipopigmentada, vesiculoampollosa, palmoplantar, pustular e incluso la denominada como MF invisible, que se manifiesta en forma de prurito sin lesiones cutáneas. La entidad conocida como piel laxa granulomatosa es considerada, en la clasificación de la EORTC para linfomas cutáneos primarios, como un linfoma cutáneo de células T encuadrado en el apartado provisional, habiéndose descrito su asociación con MF clásica y enfermedad de Hodgkin.
Manifestaciones extracutáneas.- En las fases avanzadas la MF puede mostrar manifestaciones extracutáneas, principalmente linfadenopatías, y posteriormente afectación de órganos internos, como hígado, bazo, pulmón, tracto gastrointestinal y médula ósea. Con frecuencia las lesiones viscerales son asintomáticas o infravaloradas, ante lo llamativo del cuadro cutáneo, y se trata en muchos casos de hallazgos de autopsia. En general, la afectación visceral se observa en pacientes con lesiones cutáneas avanzadas, adenopatías y células de Sézary circulantes.
-Síndrome de Sézary
El SS se define por la tríada de eritrodermia, linfadenopatías generalizadas y presencia de células T neoplásicas en piel, ganglios linfáticos y sangre periférica. No está aún bien determinado el número de células atípicas circulantes que son necesarias para definir el síndrome. El Grupo de Estudio del Linfoma Cutáneo de la EORTC sugirió como criterio adicional la demostración en sangre periférica de células T clonales y la presencia de una población elevada de células T CD4+, con el consiguiente incremento de la relación CD4/CD8 (> 10).
Clínicamente se manifiesta en forma de eritrodermia y adenopatías, con edema e intenso prurito. Adicionalmente se puede encontrar hepatomegalia (57% de los casos), alopecia (32%), onicodistrofia (32%), queratodermia (29%), ectropión con inflamación ocular secundaria y en las formas severas facies leonina.
Histopatología.- En las fases iniciales de la MF el diagnóstico histológico puede ser difícil. Se caracteriza por un infiltrado linfocitario escaso en dermis papilar, con tendencia a disponerse en banda subepidérmica. Estos linfocitos presentan epidermotropismo o tendencia a invadir la epidermis.
A medida que la enfermedad progresa y las placas se van engrosando, el infiltrado se hace mayor y se dispone en banda densa en dermis papilar y a nivel de dermis reticular, adopta un patrón perivascular superficial y profundo o casi difuso. Estos linfocitos son más atípicos, de mayor tamaño y con frecuencia forman en la epidermis microabscesos de Pautrier.
Finalmente, en la fase tumoral disminuye el grado de epidermotropismo y el infiltrado tiende a hacerse más denso y profundo. En esta fase algunas células pierden la morfología cerebriforme característica y se transforman en células grandes.
Inmunohistoquímica.- Desde el punto de vista inmunohistoquímico la célula proliferante en la MF/SS es el linfocito T maduro cooperador de memoria (CD3+, CD5+, CD43+, CD45RO+, CD45+, CD4+, CD8, CD20, CD30), a diferencia de las células T maduras. Se han descrito casos aislados con fenotipo T supresor CD4 CD8+ y en las fases tardías de la MF, principalmente tumoral, puede haber inmunofenotipos aberrantes.
Diagnóstico.– El diagnóstico de la MF/SS se basa en los hallazgos clínicos combinados con la histología de la biopsia de piel. En aquellos pacientes con lesiones incipientes los hallazgos histológicos pueden ser poco específicos y en estos casos es importante realizar un seguimiento periódico del enfermo y practicar biopsias repetidas. Existen, no obstante, técnicas diagnósticas adicionales que pueden ser útiles en determinados casos o ayudar al diagnóstico precoz. Algunas de estas técnicas, como la microscopia electrónica, citometría del DNA e índice del contorno nuclear, apenas se utilizan en la práctica clínica cotidiana y han sido desplazadas en los últimos años por el diagnóstico inmunogenético.
El diagnóstico inmunogenético de la MF/SS puede realizarse con técnicas de Southern blot o de PCR (reacción en cadena de la polimerasa).
— Técnicas de Southern blot. Con esta técnica se estudia el gen que codifica la cadena beta del receptor T. Presenta algunas limitaciones como la utilización de tejido fresco o congelado y una sensibilidad limitada (sólo permite detectar una proliferación clonal cuando ésta representa entre el 1-5% del DNA total), por lo que no es útil para detectar monoclonalidad en los primeros estadios de la MF en los que el porcentaje de células neoplásicas en relación con el infiltrado benigno reactivo es mínimo.
— Reacción en cadena de la polimerasa (PCR). En la actualidad la PCR está desplazando al Shouthern blot por su mayor sencillez, sensibilidad y permitirnos manejar material incluido en parafina.
Estudio de extensión y clasificación.- Una vez realizado el diagnóstico de MF/SS se procede a practicar un estudio de extensión de la enfermedad. Para este fin se emplea un protocolo de estudio que incluye la solicitud a todos los pacientes de un hemograma y fórmula leucocitaria, determinación de células de Sézary circulantes, VSG, bioquímica hemática, enzima láctico deshidrogenasa, beta 2 microglobulina, radiografía de tórax y biopsia de piel. La mayoría de los investigadores están de acuerdo en hacer biopsia de ganglios linfáticos sólo en aquellos casos en que éstos están agrandados. El valor de la biopsia de médula ósea o el escáner abdominal y pélvico está menos determinado y debe de ser reservado para aquellos pacientes con tumores, linfocitos atípicos circulantes, afectación ganglionar o síntomas sugestivos.
Conocida la extensión de la enfermedad se clasifica a los pacientes en distintos estadios. El sistema de clasificación más empleado es el sistema TNM, introducido en 1979 por el National Cáncer Institute y modificado en 1988 por Sausville y cols.
Transformación a linfoma de célula grande (lcg)
Algunos pacientes con MF/SS desarrollan a lo largo de su evolución un cambio en la morfología celular, con transformación de las células de pequeño o mediano tamaño en células grandes. En general se acepta que cuando las células grandes constituyen más del 25% del infiltrado o bien forman nódulos microscópicos, la MF/SS se ha transformado a LCG. Con frecuencia se acompaña de una evolución más agresiva de la enfermedad y supervivencia mediana más corta.
Tratamiento.- En la actualidad no existe un único tratamiento para la MF, sino que nos encontramos ante distintas alternativas terapéuticas cuyo objetivo principal es ofrecer un aumento en las expectativas de vida, así como una mejora de su calidad. El tratamiento actual de la MF incluye, principalmente, las siguientes posibilidades:
- –Corticoides tópicos
- Pueden ser útiles en fases iniciales, aunque generalmente la respuesta es incompleta y de corta duración.
- -Quimioterapia tópica: La quimioterapia tópica está indicada principalmente para el tratamiento de la MF en placas no infiltradas o que presentan una infiltración ligera o moderada. Disponemos de dos sustancias: mecloretamina (actualmente no comercializada) y carmustina.
Tratamientos que han dado resultados satisfactorios:
- -Ultravioleta B
- -Fotoquimioterapia (PUVA)
- -Retinoides orales
- -Retinoides orales más PUVA (RePUVA)
- -Fotoquimioterapia extracorpórea
- -Interferón alfa
- -Baño de electrones (electron beam therapy)
- -Radioterapia convencional de ortovoltaje
Puede ser efectiva para erradicar tumores cutáneos aislados
- -Quimioterapia sistémica con metotrexate dependiendo del estadiaje.
Bibliografía
- Sézary A, Bouvrain Y. Erythrodermie avec présence des cellules monstrueuses dans le derme et le sang circulant. Bull Soc Fr Derm Syphiligr 1938;45:254-60.
- Mycosis fungoides and Sezary syndrome. Blood 1996;88:2385-409.
- Updated kiel classification for lymphomas. Lancet 1988;1:292-3.
- National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: aummary and sescription of a working formulation for clínicasl usage. Cancer 1982;49:2112-35.
- A revised european-american classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994;84:1361-92.
- World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the clínicasl advisory committee meeting-airlie house, Virginia, november 1997. J Clin Oncol 1999;17:3835-49.
- EORTC classification for primary cutaneous lymphomas: a proposal from the cutaneous lymphoma study group of the european organization for research and treatment of cancer. Blood 1997;90:354-71.
- Cutaneous T-cell lymphoma (mycosis fungoides). Lancet 1996;347:871-6.
- Mycosis fungoides in the United States. Increasing incidence and descriptive epidemiology. JAMA 1988;260:42-6.
- Alibert JLM, ed. Tableau du plan fungoide. Description des maladies de la peau observées a l’Hopital St. Louis. Paris: Barroisl’Aine et fils, 1806;157. (Cit en: MacKie RM. Lymphomas and leukaemias. En: Champion RH, Burton JL, Ebling FJG, eds. Rook/ Wilkinson/ Ebling Textbook of Dermatology, 5.ª ed. Oxford: Blackwell Scientific Publ; 1992:2107-34).
- Weinstock MA, Horm JW. Mycosis fungoides in the United States. Increasing incidence and descriptive epidemiology. JAMA 1988;260:42-6.
- Ketron LW, Goodman MH. Multiple lesions of the skin apparently of epithelial origin resembling clínicaslly micosis fungoides. Arch Derm Syph 1931;24:758-77.
- Verret JL, Rousselet MC, Peria P. Mycosis fongoïde unilésionnel en plaque. 3 observations. Ann Dermatol Venereol. 1997;124:527-30.
- Sausville EA, Eddy JL, Makuch RW, y cols. Histopathologic staging at initial diagnosis of mycosis fungoides and Sézary syndrome. Definition of three distinctive prognostic groups. Ann Intern Med 1988;109:372-82.
- Epidemiology of mycosis fungoides. Semin Dermatol 1994;13;154-9.
- Navas IC, Ortiz-Romero PL, Villuendas R y cols. P16INK4a gene alterations are frequent in lesions of mycosis fungoides. Am J Pathol 2000; 156: 1565-157
- No detection of HTLV-I proviral DNA in lesional skin biopsies from swiss and german patients with cutaneous T-cell lymphoma. Br J Dermatol 1996;134:282-4.
- Mycosis fungoides a disease of antigen persistence. Br J Dermatol 1974;91:607-16.
- p16 ink4a gene alterations are frequent in lesions of mycosis fungoides. Am J Pathol 2000;156:1565-72.
- Mycosis fongoide. En: Degos R, ed. Dermatologie. Paris: Flammarion; 1981:897-910a.
- Woringer-Kolopp disease. Pagetoid reticulosis. Dermatologica 1978;156:283-91.
- Mycosis fongoide de unilesionnel en plaque. 3 observations. Ann Dermatol Venereol 1997;124:527-30.
- Lymphomes cutans par des capillarites purpuriques et pigmentaires. Ann Dermato Venereol 1999;126:321-6.
- Histopathologic staging at initial diagnosis of mycosis fungoides and Sezary syndrome. Definition of three distinctive prognostic groups. Ann Intern Med 1988;109:372-82.
- Linfomas cutáneos. Med Cutan Iber Lat Am 1998; XXVI:113-36.
- Topical corticosteroids for mycosis fungoides. Experience in 79 patients. Arch Dermatol 1998;134:949-54.
- Treatment of cutaneous T-cell lymphoma. Semin Dermatol 1994; 13:207-15.
- Cutaneous T cell lymphoma: update of treatment. Dermatology 1999;199:102-5.
- Topical treatment of early cutaneous T-cell lymphoma. Hematol Oncol Clin North Am 1995;9:1031-56.
- Topical carmustine (BCNU) for patch/plaque mycosis fungoides. Semin Dermatol 1994;13:202-6.
- Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol 1992;128:931-3.
- Retinoids plus PUVA (RePUVA) and PUVA in mycosis fungoides, plaque stage. A report from the Scandinavian Mycosis Fungoides Group. Acta Derm Venereol (Stockh) 1989;69:536-8.
- Chemotherapy for mycosis fungoides and the Sezary syndrome. Hematol Oncol Clin North Am 1995;9:1109-16.
- Santiago Gómez Díez, Narciso Pérez Oliva. Micosis fungoide y síndrome de Sézary. Actas Dermosifiliogr 2001;92:193-206