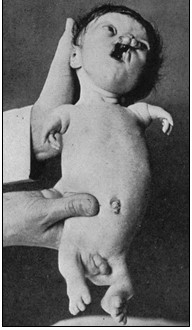
INFORMACIÓN BÁSICA.- Síndrome de Roberts (SR) (Focomelia Pseudotalidomídica) (ORPHA:3103/CIE-10:Q73.8/OMIM:268300). El SR o Focomelia (FM). Ambas son expresión de una misma intensidad, aunque de intensidad variable. El primero se caracteriza por retraso en el crecimiento pre- y postnatal, anomalías graves y simétricas de los miembros, malformaciones craneofaciales y déficit intelectual grave. La FM es una forma moderada del SR, estado que posee mi paciente.
El SR es una patología de origen genético con patrón autosómico recesivo, que cursa con malformaciones congénitas múltiples y una separación prematura de la heterocromatina centromérica de los cromosomas en el 50% de los casos.
Como cualquier otra malformación, es consecuencia de alteraciones en el desarrollo embrionario; las causas pueden ser genéticas o ambientales. En los años sesenta del siglo pasado, su incidencia aumentó de forma espectacular debido a la prescripción de talidomida para aliviar las náuseas del embarazo. Recientemente se han identificado una molécula a la que la talidomida se une tras lo que modifica su actividad. Es conocido que esta molécula, si no funciona bien, afecta al funcionamiento de factores de crecimiento vascular durante el desarrollo fetal, lo que podría explicar los efectos perniciosos de la talidomida.
Aunque queda por descartar si existe la posibilidad de que la madre de mi paciente ingirió durante su embarazo otros medicamentos, o tuvo relaciones endogámicas que ocasionaron la mutación descrita, y que pueden ocasionar reducción en el desarrollo embrionario de las extremidades; entre los fármacos responsables se citan los siguientes: etreninato, itraconazaol, ketoconazol, metronidazol, propoxifeno, quinina, rifampicina, fluconazol, hiocinamina, atropina, azatadina, ácido valproico, ciclosporina, ducumarínicos, disulfiram, etc., o focomelia per se (disulfiram, indometacina, sumatriptan, trifluoperacina).
Los investigadores han producido FM con rayos X en embriones de pollo y han llevado a cabo un análisis molecular y de linaje celular para ver cómo se produce la enfermedad. Hasta el momento, se creía que la enfermedad se originaba según el modelo de la ‘zona de progreso’: al reducirse el tamaño de la extremidad debido a la pérdida de células (por la muerte causada, en este caso, por la irradiación), las células supervivientes permanecerían más tiempo en la zona de progreso, adquiriendo destinos cada vez más distales (alejados del centro del cuerpo). Es decir, que las células que normalmente darían lugar al húmero acababan formando los dedos.
Se pueden observar otras malformaciones estructurales en niños expuestos al fármaco en los dos primeros meses de vida intrauterina: hemangioma facial, atresia esofágica y duodenal, tetralogía de Fallot, agenesia renal y anomalías del oído externo (anotia o microtia).
El síndrome focomelia tipo Schinzel, también llamado síndrome hipoplasia/aplasia de la/s extremidades/pelvis, se caracteriza por malformaciones esqueléticas que afectan al cúbito, huesos pélvicos, peroné y al fémur. Debido a que el fenotipo es similar al síndrome de malformación Al-Awadi/Raas-Rothschild, se cree que pueda tratarse del mismo trastorno. Solamente se han descrito algunos casos. Los pacientes tienen malformaciones intercalares de los miembros (focomelia combinada a veces con polidactilia, oligodactilia o ectrodactilia), ausencia o hipoplasia de huesos pélvicos (incluyendo agenesia sacral o hipoplasia), defectos del cráneo (con frecuencia, defecto del hueso occipital con o sin meningocele).
Prevalencia.- La prevalencia y la incidencia son desconocidas: menos de 150 casos aparecen descritos en la literatura al momento. En general las anomalías de las extremidades afectan a 6 de cada 10.000 nacidos vivos y son más frecuentes en extremidades superiores que en las inferiores. En los EE. UU., se estima que esta enfermedad es menos de 5,000. En Ecuador desconocemos su prevalencia.
Transmisión.- Autosómico recesivo.
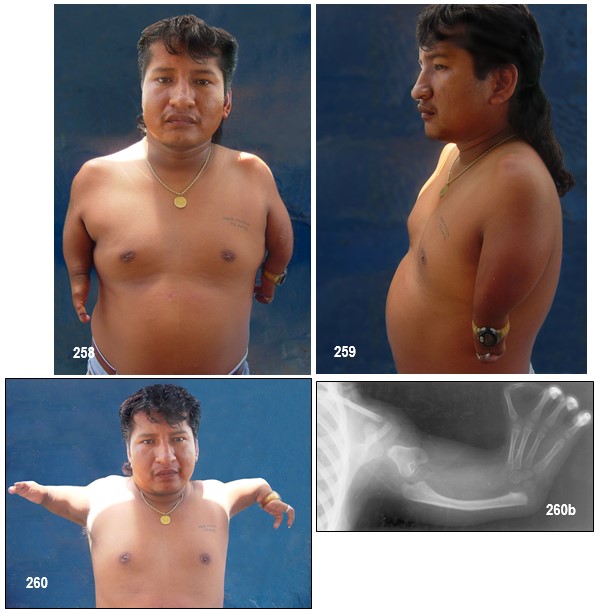
Manifestaciones clínicas.- La FM es una malformación de las extremidades muy rara. Se caracteriza por un acortamiento de los elementos de la extremidad más cercanos al cuerpo; los elementos más alejados, los dedos, quedan menos afectados (como se observa en el paciente de la imagen). En casos graves, manos, pies o incluso dedos surgen directamente del tronco, sin rastro de los elementos intermedios. Las extremidades superiores están afectadas con mayor frecuencia y gravedad que las extremidades inferiores. La afectación es esencialmente mesomélica, predominante en el radio en las extremidades superiores y en el peroné en los miembros inferiores. La afectación más grave resulta en una focomelia. Los pulgares aplásicos o hipoplásicos, la oligodactilia, la clinodactilia o la sindactilia también pueden estar presentes. Las anomalías craneofaciales incluyen: microcefalia (más grave en hombres que en mujeres), alas nasales hipoplásicas, hipoplasia malar, hipertelorismo, micrognatia, hemangioma capilar, exoftalmos, fisuras palpebrales con inclinación hacia abajo, orejas displásicas o pequeñas, córnea opaca o cataratas y fisura labio-palatina.
La gravedad de las anomalías craneofaciales está correlacionada con el grado de afectación de las extremidades. Pueden presentarse otras malformaciones: anomalías congénitas de corazón, riñón quístico, o genitales agrandados (pene o clítoris grandes).
Otras características adicionales pueden incluir distrofia torácica, facies inusual (orejas displásicas y grandes, así como paladar alto y estrecho), y malformaciones en los genitales (aplasia mulleriana, agenesia del útero y vagina, micropene con criptorquidismo). El crecimiento y el desarrollo mental son normales. Debido a que la mayor parte de los pacientes afectados son hijos de padres consanguíneos, se presume que pueda tratarse de una herencia autosómica recesiva. Mutaciones inactivantes del gen WNT7A (3p25) son las responsables de este síndrome. Se trata de una forma alélica grave del síndrome de Fuhrmann. Las malformaciones de las extremidades y/o la ausencia de pelvis se pueden detectar por ecografía. Algunos de los niños afectados murieron poco después de nacer.
Se observa déficit intelectual grave en pacientes que sobreviven al periodo neonatal.
Genética.- La afección puede heredarse u ocurrir esporádicamente. También está relacionado con la exposición prenatal al fármaco contra las náuseas talidomida. En el síndrome de Holt-Oram, las formas más graves se manifiestan con focomelia con extremidades rudimentarias. La enfermedad está causada por mutaciones en el gen ESCO2 (8p21.1/ pares de bases 27.774.540 a 27.812.623). Este gen codifica para una proteína perteneciente a la familia Eco1/Ctf7 de acetiltranferasas y que participa en la cohesión de cromátidas hermanas durante la fase S.
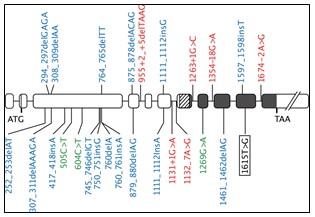
El gen ESCO2 proporciona instrucciones para hacer una proteína que es importante para la separación de los cromosomas durante la división celular adecuada. Antes de las células se dividen, deben copiar todos sus cromosomas. El ADN copiado de cada cromosoma se arregla en dos estructuras idénticas, llamado cromátides hermanas. La proteína ESCO2 juega un papel importante en el establecimiento del pegamento que mantiene las cromátides juntos hasta que los cromosomas están dispuestos a separar.
Se han encontrado al menos 26 mutaciones para causar el SR. Todas estas mutaciones evitan que la célula pueda producir cualquier proteína ESCO2 funcional, ESCO2 provocan retraso de la división celular, incremento de la muerte celular y defectos en la proliferación celular. La pérdida de las células progenitoras durante la embriogénesis podría ser responsable de las anomalías del desarrollo observadas en el SR. Algunas mutaciones cambian los bloques de construcción de proteínas individuales (aminoácidos), mientras que otros dan lugar a una proteína anormalmente corto. La ausencia de la proteína ESCO2 funcional hace que algunos de la cola entre las cromátidas hermanas que falta alrededor del punto de constricción de los cromosomas (centrómero). En el SR, las células responden a anormal unión de cromátidas hermanas al retrasar la división celular. División celular tardía puede ser una señal de que la célula debe someterse a la auto-destrucción. Los signos y síntomas del SR pueden ser debido a la pérdida de células de diversos tejidos durante el desarrollo temprano. Los investigadores originalmente sospechaban que la gravedad variable de SR fue causado por diferentes tipos de mutaciones en el ESCO2 gen. Se predijo que las personas con la forma leve de la enfermedad podrían tener mutaciones que reducen la actividad de la proteína ESCO2, mientras que aquellos con la forma grave tendrían mutaciones que eliminan por completo la función de la proteína. Sin embargo, todas las mutaciones conocidas en elESCO2 gen impiden la producción de cualquier proteína ESCO2 funcional. La causa subyacente de la variación en la gravedad de la enfermedad sigue siendo desconocida. Los investigadores sospechan que otros factores genéticos y ambientales pueden estar implicados.
Clasificación de la focomelia.- Como se definió anteriormente es la ausencia de los huesos largos del miembro superior a veces la mano aparece unida al tronco y en otras oportunidades, el húmero es solo el ausente y el antebrazo se encuentra unido al tronco (focomelia proximal), también el antebrazo puede estar ausente y en ese caso la mano se une al brazo (focomelia distal).
La focomelia típica, cuando es total afecta los cuatro miembros (tetrafocomelia) y cuando es parcial a uno de los dos de los de estos.
La focomelia puede clasificarse utilizando el sistema de Frantz y O’Rahilly.
Las extremidades inclasificables se dividen en tres grupos distintos:
- Tipo A: un húmero anormal con una anomalía hueso único del antebrazo
- Tipo B: un húmero anormal con radio anormal y cúbito.
- Tipo C: un húmero anormal fusionado a un antebrazo hueso o huesos.
Ha veces parecía no haber diferencias significativas entre los patrones de focomelia en los afectados por la talidomida y los que no habían sido expuesto a esta droga.
Diagnóstico.- Este se basa en los signos clínicos y en el cariotipo (apariencia típica de los cromosomas en forma de «rieles de tranvía», debido a la repulsión de las regiones heterocromáticas y a la separación prematura del centrómero).
La secuenciación directa del gen ESCO2 puede confirmar el diagnóstico. El diagnóstico prenatal puede producirse accidentalmente durante una ecografía prenatal rutinaria en el 3º trimestre.
En embarazos de alto riesgo, o en aquellos en los que se sospeche acondroplasia tras la ecografía, puede estudiarse el ADN fetal para la mutación FGFR3 para confirmar el diagnóstico.
Diagnóstico diferencial.- incluye la embriopatía por talidomida y los síndromes de Baller-Gerold, Cornelia de Lange y TAR (ver estos términos). Además el síndrome focomelia tipo Schinzel, la hipocondroplasia, el enanismo tanatofórico (tipos I y II), y SADDAN (véase Displasias óseas, en este mismo capítulo). Para las familias con un hijo afectado de SR y portador de mutaciones ESCO2 (riesgo de recurrencia del 25%), se puede proponer un diagnóstico prenatal mediante el análisis de DNA de las muestras de vellosidades coriónicas. En los casos de sospecha del SR por observación de las anomalías características de SR en ecografía, el diagnóstico puede confirmarse mediante el cariotipo.
Tratamiento.- El manejo incluye: cirugía correctiva de las anomalías faciales, tratamiento quirúrgico y/o ortopédico de las malformaciones de las extremidades y el manejo de las discapacidades cognitivas. El pronóstico es relativamente desfavorable. La alta mortalidad en el período neonatal o durante la primera infancia se debe a las malformaciones cardíacas o renales.
Hechos históricos.-
Bibliografía
- http://www.orpha.net
- Roberts JB. A child with double cleft of lip and palate, protrusion of the intermaxillary portion of upper jaw and imperfect development of the bones of the four extremities. Ann Surg. 1919;70:252-4.
- https://rarediseases.info.nih.gov/diseases/12123/phocomelia
- Dra. Miriam Sánchez-Segura, Dra. Vianed Marsán–Suárez, DraC. Consuelo Macías-Abraham, Dra. Norma de León-Ojeda, Dra. Alina García-García, Dra. Isabel Toledo-Rodríguez, Lic. Lázaro del Valle-Pérez, Lic. Berta Beatriz Socarrás–Ferrer. Síndrome de Roberts asociado con inmunodeficiencia. Rev Cubana Hematol Inmunol Hemoter vol.28 no.2 Ciudad de la Habana abr.-jun. 2012
- Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo I, 2018, UEES, Ecuador.