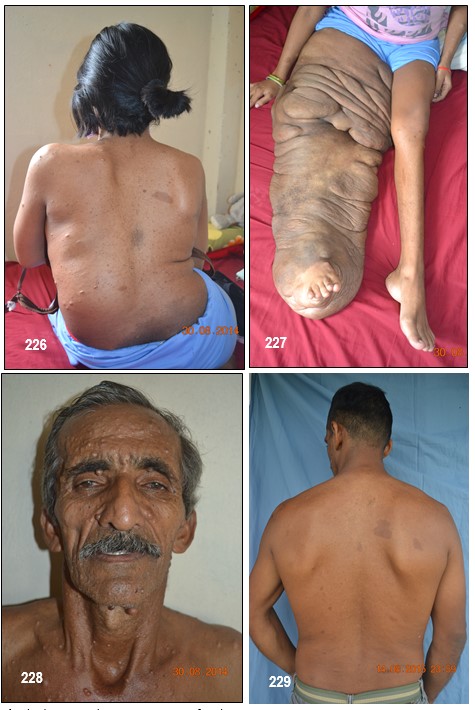
Comentarios de los Editores: A continuación describimos seis pacientes de una misma familia con NF-tipo 1, en 3 generaciones. El primer caso, madre de la segunda paciente, abuela del tercero, cuarto, y quinto caso respectivamente. La presentación de estos casos certifica que la NF-1 se hereda con rasgos autonómico dominante.
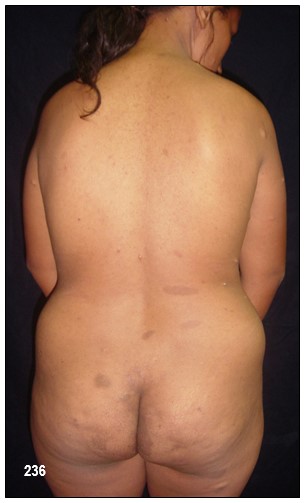
El término neurofibroma literalmente significa un tumor de tejidos derivados de células nerviosas (neuro) y tejido fibroso (fibroma). Los neurofibromas plexiformes involucran a nervios profundos, grandes y muestran fascículos irregulares como consecuencia del incremento de la matriz endoneural y del perineuro, sin aumento del número de fibras nerviosas.
INFORMACION BÁSICA: Neurofibromatosis.- Neurofibromatosis de Tipo 1 (NF-1) (ORPHA:636/CIE-10:q85.0/OMIM:162200-166210-613675).- Esta condición clínica (básicamente un síndrome neurocutáneo) presenta numerosas y tenues, a veces graves, alteraciones en infinidad de partes del cuerpo, especialmente en los nervios periféricos, y a distintos niveles y grados de compromiso.
La NF-1 es una enfermedad genética progresiva de heterogeneidad y de gran variabilidad clínica. Esta patología puede manifestarse de muchas maneras diferentes en muchos tejidos diferentes dentro del mismo paciente y entre los miembros familiares. La NF-1 es un desorden del sistema del neuroectodermal que produce tumores hamartomatosos benignos de cualquier órgano o sistema (el más notablemente la piel, los ojos, y el sistema nervioso) ese aumento en el número clasifica según el tamaño a lo largo de la vida. Estos tumores son de tejidos derivados de la cresta neural, nervios particularmente sensorios, células de Schwann, y melanocitos.
Esta dolencia es altamente penetrante; heredándose de manera autosómica dominante, es decir, la probabilidad que tiene un afectado en desarrollar el fenotipo o cualquier manifestación de la enfermedad asociada con aquel. Un 50% son hereditarios y la otra mitad por mutaciones espontaneas. Dicho esto, hay que decir que el 50% de los pacientes afectos presentarán una forma leve de la enfermedad, y 1/3 desarrollarán complicaciones potencialmente graves. Entre un 30-60% de los pacientes manifiestan trastornos del aprendizaje, y en un 4-8% el CI es < 70. En un 2% pueden presentar hipertensión arterial por estenosis de la arteria renal. Otras posibles complicaciones son ortopédicas (escoliosis, arqueamiento de huesos largos), oftalmológicas (glioma óptico en el 15%), endocrinológicas (talla corta, feocromocitoma), etc.
El diagnóstico de la NF-1 se basa en criterios clínicos establecidos. Hasta el momento, los estudios realizados han encontrado una gran variedad de mutaciones genéticas puntuales, pero aun no ha sido identificada una mutación frecuentemente recurrente. Las investigaciones tampoco han hallado una relación entre una mutación particular y sus consecuentes manifestaciones clínicas. Esta variabilidad sugiere la presencia de otros factores aún no determinados para aclarar dichas manifestaciones.
Es altamente reconfortante el saber acerca de la gran cantidad de investigaciones médicas 3actuales tendientes a descubrir nuevos tratamientos para este malestar genético.
La NF-1 se puede clasificar en cuatro grados: mínima, suave, moderada y severa, pero se desaconseja esta práctica debido a la extrema variabilidad del desorden genético. El rango en su severidad varía desde casos demasiado tenues o mínimos en donde solamente se presentan algunas manchas cafés con leche, (MCL), y unos cuantos tumores (neurofibromas) hasta circunstancias altamente severas en las cuales aparecen todas las características aquí descritas. Hasta la fecha, no se ha podido determinar la forma de su desarrollo pues esta anomalía, sea cual sea su estado, es susceptible siempre de empeorar y nunca de mejorar. En algunas ocasiones, los síntomas, ya sean tenues o graves, no son claramente identificados. Estos se presentan al nacer o se desarrollan durante la niñez o la adolescencia. Sin embargo, existe la posibilidad de no manifestarse tardíamente. Es poco probable que quienes tengan NF-1 y lleguen a la edad adulta sin presentar problemas del tipo grave, estos se desarrollen en un futuro. En la gran mayoría de los casos de NF-1, un diagnóstico preciso se puede indicar a los 8 años de edad del paciente y la totalidad a los 20
El 33% de los afectados con este síndrome neurocutáneo son asintomáticos e identificados durante controles rutinarios, 33% presentan problemas cosméticos y 33% neurológicos.
Aproximadamente el 60% de los afectados la padecen en grado mínimo y pueden llevar unas vidas muy productivas; un 20% tiene problemas corregibles y un 20% desarrollan dificultades persistentes. El 7% de las personas con NF-1 se considera en riesgo de padecer de las condiciones graves. Sus manifestaciones dependen primordialmente de la edad del paciente. Se han determinado dos rangos para las manifestaciones severas. El primero entre los 0 y los 10 años y el segundo entre los 36 y los 50 años, período en el cual pueden surgir tumores malignos. Esto ocurre en el 5% de las veces, pero no es común. Se cree que la esperanza de vida de los afectados se reduce entre 10 y 15 años del promedio de la gente.
Las causas de mayor mortandad son la hipertensión y el cáncer.
Del total estudiado por el cuerpo médico, un 64% corresponde a casos entre moderados y severos.
Por lo general, solamente las personas con manifestaciones graves o por aspectos estéticos, son quienes acuden a consultas médicas. Quienes tienen pocos problemas, le restan importancia.
La NF-1 no incapacita a la gran mayoría de los afectados y ellos pueden perfectamente continuar una vida activa normal. De presentarse alguna sintomatología extraña en los tumores, ella podría desencadenar una incapacidad dependiendo del compromiso.
La ciencia se inclina a estudiar únicamente los casos graves. Por cada uno de ellos, hay miles desapercibidos. Los casos severos han sido catalogados por esta razón más no por la característica principal del trastorno genético, ya sea neurológica u ortopédica.
Hasta el cierre de esta revisión, se desconoce si la NF proviene de causas relacionadas con el medio ambiente.
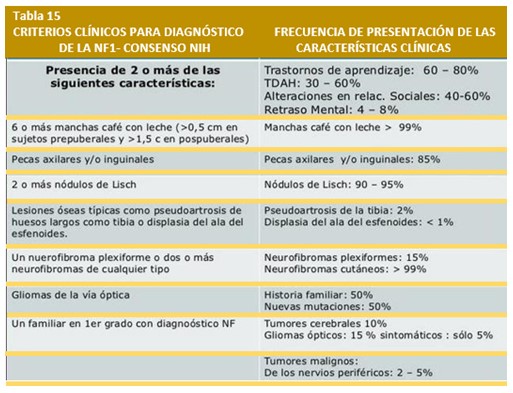
La NF-1 se diagnostica cuando se manifiestan dos o más de las siguientes alteraciones corporales siempre y cuando no haya sido otra la enfermedad diagnosticada. Estos pueden ocurrir a cualquier edad, algunos de ellos se presentan a edades específicas y algunas de las manifestaciones son el resultado de complicaciones de la lesión original:
Prevalencia.- La enfermedad afecta a uno de cada 3000 individuos y se presenta en uno de cada 200 pacientes con retraso mental. Casi el 50 % de los casos representan neomutaciones.
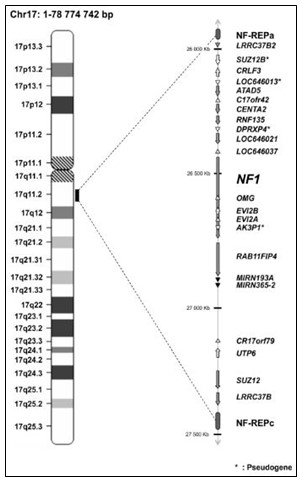
Patogénesis molecular.- La NF-1 es uno de los desórdenes genéticos heredables más comunes en el hombre. Su causa proviene de diferentes mutaciones en un complejo gen conocido como NF-1: un defecto en el cromosoma 17 en su sección q11.2 dando como resultado un menor o nulo desempeño en el organismo de la proteína neurofibromina. La NF1 es una enfermedad muy variable y no hay dos personas afectadas de la misma manera, inclusive aunque sean de la misma familia.
El gen de la NF-1, causante de la enfermedad, se localiza en la región pericentromérica del brazo largo del cromosoma 17, en 17q11.2, y se expresa en el SNC, SNP y medula espinal. Es un gen de gran tamaño, de 290 kilobases y 60 exones. Forma parte de la familia de genes supresores tumorales y codifica la síntesis de una proteína citoplasmática, la neurofibromina (de unos 2.818 aminoácidos y de 220 kDa) expresada normalmente en diferentes células del sistema nervioso (neuronas, oligodendrocitos y células de Schwann), queratinocitos y melanocitos.
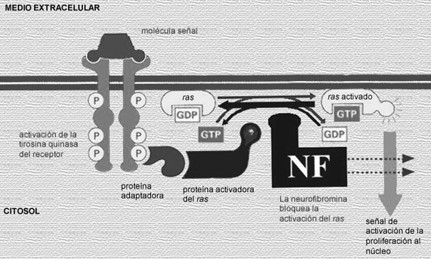
La mayoría de las mutaciones de este gen dan lugar a la síntesis de una proteína truncada (esta proteina es supresora de tumores y de la proliferación celular), y un 5% de estas mutaciones son grandes deleciones. Las mutaciones de NF-1 suelen ocasionar una disminución de los valores intracelulares de neurofibromina, lo cual parece suficiente para producir la mayoría de las manifestaciones clínicas de la enfermedad. La neurofibromina presenta semejanzas estructurales con las proteínas GAP de los mamíferos. Esta familia de genes está implicada en el control, el crecimiento y la diferenciación celular mediante su interacción con la familia de los genes Ras.
La neurofibromina tiene un dominio homólogo a la guanosintrifosfatasa (GTPasa) y actúa activando la familia de las proteínas GAP. La actividad GAP acelera la hidrólisis de la GTP unida a Ras, convirtiendo la proteína p21ras de forma activa a inactiva (GDP). Esta homología funcional sugiere que la neurofibromina regularía el crecimiento inhibiendo la señal mediada por Ras. Como consecuencia de esta inhibición, existiría una alteración del crecimiento, la proliferación celular, el desarrollo del aprendizaje, la memoria y la plasticidad sináptica.
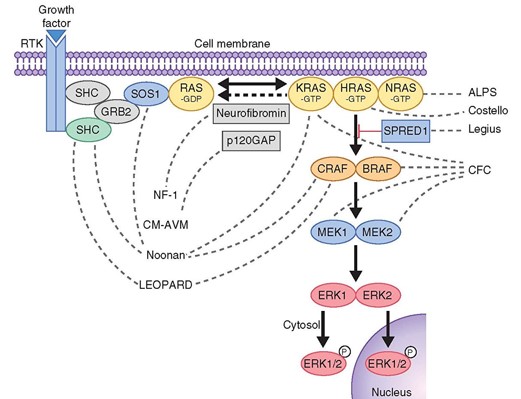
Además de regular la actividad de Ras, la neurofibromina potencia la actividad de la adenilciclasa estimulada por la activación de la proteína-G y de los receptores neuropeptídicos, que desempeñan un papel en el aprendizaje y en la memoria. En algunos modelos animales con alteración del gen de la NF-1 se ha evidenciado un aumento de la susceptibilidad para el desarrollo de neoplasias, lo que apoya el posible papel del gen de la NF-1 como gen supresor tumoral. Los tumores en la NF invariablemente presentan células deficientes de neurofibromina
Diversos os estudios realizados en tumores desarrollados en NF-1 han confirmado la presencia de una pérdida de heterocigosidad para el gen de la NF-1 11. Se ha demostrado que es necesaria la doble inactivación del gen de la NF-1 en las células de Schwann para el desarrollo de neurofibromas. Las células de Schwann deficientes en neurofibromina secretan una sustancia que estimula la migración de los mastocitos, que facilitarían la producción de la matriz extracelular y de la angiogénesis. Bajenaru et al 14 observaron en estudios experimentales con ratones que los astrocitos deficitarios en la expresión de NF-1 no son capaces de desarrollar gliomas del nervio óptico.
Manifestaciones clínicas.– El instituto Nacional de la Salud de EE.UU, definió en 1987, los criterios de la NF-1 20. Para el diagnóstico de neurofibromatosis, un paciente debe tener al menos 2 de los siguientes 7 criterios:
- 6 ó más manchas “café con leche”, iguales o mayores de 5 mm en pacientes prepúberes y de 15 mm de diámetro en pacientes postpúberes
- 2 o más neurofibromas de cualquier tipo, o uno plexiforme
- Presencia de pecas en axilas o ingles
- Glioma del nervio óptico
- 2 o más nódulos de Lisch (hamartomas del iris)
- Lesión ósea definida como displasia del esfenoides o adelgazamiento de la cortical de los huesos largos con o sin pseudoartrosis.
- Un familiar de primer grado afecto (padre, hermano o hijo) de acuerdo con los criterios previos.
En todo caso, el diagnostico de esta enfermedad es clínico. En ocasiones es difícil, sobre todo en los casos de niños sin antecedentes familiares Generalmente los niños suelen presentar manchas color café con leche como manifestación principal. Los neurofibromas suelen aparecer después de la pubertad. Algunos pacientes presentan arqueamiento tibial debido a una pseudoartrosis tibial. Los nódulos de Lisch, solo los presentan el 30 % de los niños menores de 6 años y la mayoría de los pacientes mayores de 20 años.
De todas formas lo que sí debemos recordar es que existe una expresividad muy variable, que da lugar a una gran variabilidad clínica entre los pacientes incluso dentro de la misma familia. Pasare a describir cada una de estas manifestaciones en detalles:
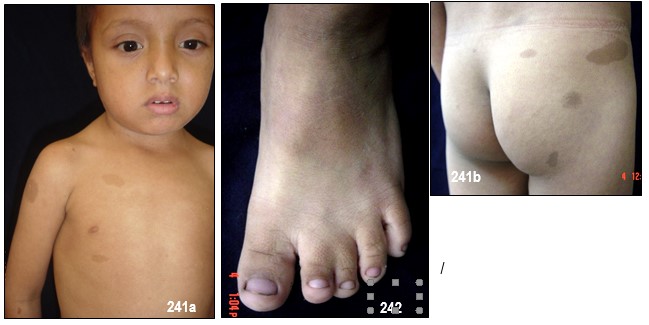
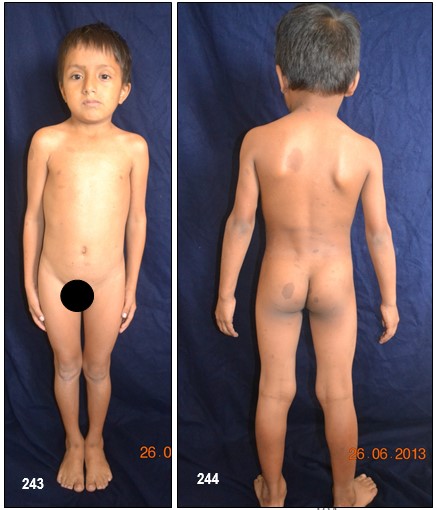
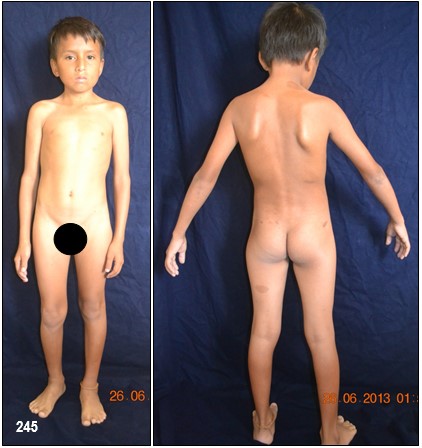
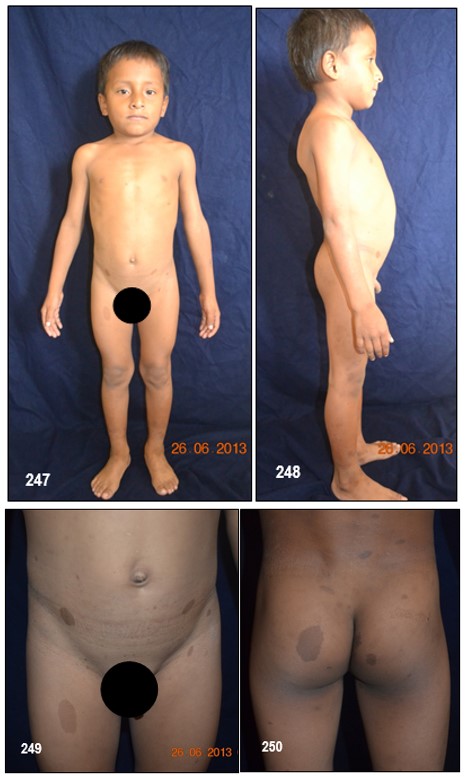
Manchas en la piel.- Alrededor del 10% de la población mundial tiene una o dos manchas color café con leche (MCL) sin padecer de NF.
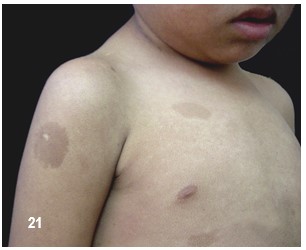
Las MCL, son áreas o puntos planos no malignos e inocuos de pigmentación sobre la piel. El 90%, si no el 100%, de los afectados con NF-1 las padecen. Su color es uniforme y se parece, como su nombre lo indica, al color del café con leche. A veces, estas son tan pálidas que pasan desapercibidas durante un examen médico. En las pacientes con piel oscura, estas manchas aparecen con color más oscuro, haciéndose más difícil identificarlas.
Generalmente, las MCL son la primera manifestación en la mayoría de los niños con NF-1 (84 a 100%) y ya están presentes al momento de nacer o aparecen alrededor de los dos años de edad y se desarrollan y se incrementan en número durante la infancia y ocasionalmente más tarde asemejándose a unas pecas. Típicamente tienen de 10 a 30 mm de diámetro, son ovoides, de color uniforme y bordes bien definidos, se pueden encontrar en cualquier parte del cuerpo excepto en el cráneo, párpados, palmas y plantas. Sin embargo, existe información de que estas manchas pueden aparecer en esos lugares. Su color se va tornando más oscuro durante el crecimiento del paciente. Por regla general, los médicos consideran, que un niño, con seis o más de este tipo de manchas, cada una con un diámetro de 0,5 cm o de 1.5 cm y mayores en adultos, pueden padecer de NF-1. No existe relación alguna entre la cantidad de manchas – a veces cientos – y el grado de severidad del desorden genético. De vez en cuando, a la edad adulta, las MCL desaparecen.
El médico debe tener mucho cuidado al expresar su opinión al encontrar en un paciente MCL sin ningún otro de los síntomas descritos, salvo que existan antecedentes familiares de NF, pues puede estar ante la presencia de un extraño caso de enfermedad pigmentaria autosómica y dominante totalmente diferente a la NF-1; sin embargo, esta situación es extremadamente rara.
Estas manchas se pueden reconocer como pertenecientes a una persona con NF cuando se evidencia un suave borde y un color café claro. En las otras enfermedades, su color es más oscuro. Entre dichos males se encuentra el síndrome LEOPARD, enfermedad autosómica dominante caracterizada por múltiples manchas lentiginosas, anormalidades genitales, pulmonares, cardíacas y auditivas.
No es necesario llevar un registro pormenorizado de cada MCL. Sin embargo, sea cual fuere el tipo de pigmentación presentada en un organismo, todo nuevo cambio debe controlarse.
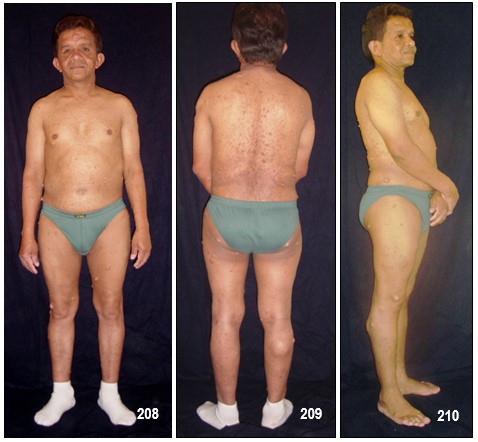
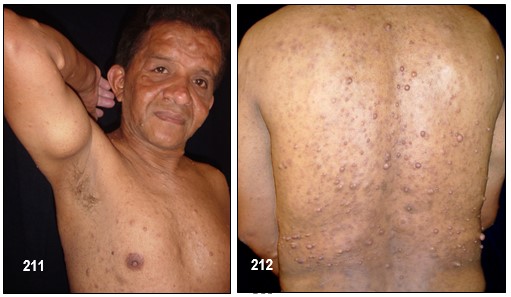
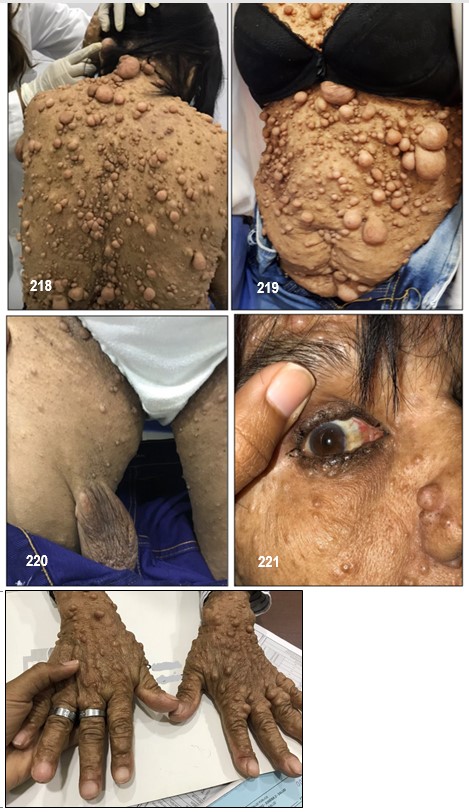
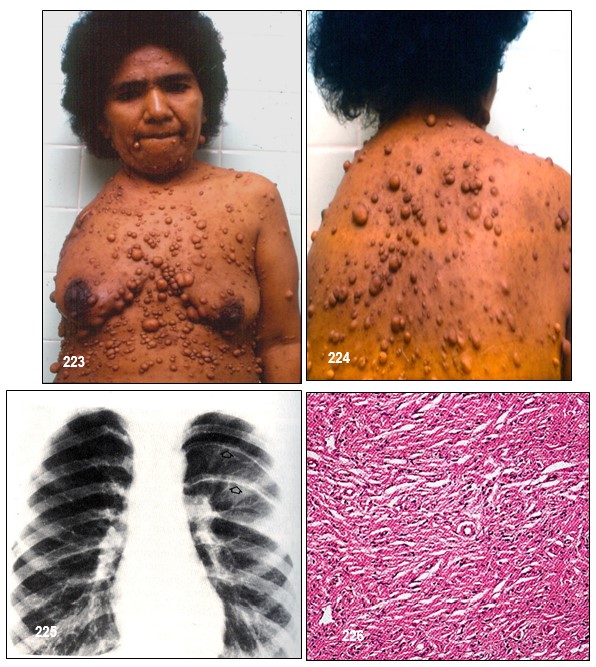
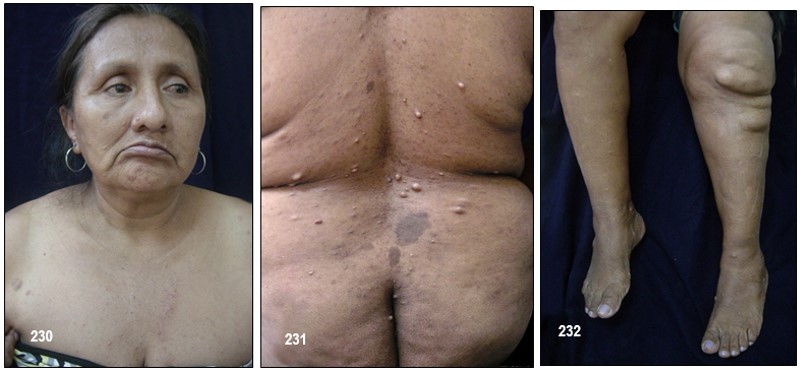
Neurofibromas.- La NF-1 puede causar miles de lesiones, a veces dolorosas, en la piel las cuales eventualmente pueden llegar a producir deformaciones y tumores periféricos benignos o verrugas sobre o debajo de la piel llamadas neurofibromas. (Neuro: tejido del sistema nervioso Fibroma: tejido fibroso). Están compuestos de células Schwann y de fibroblastos. En algunos casos aislados, tales neurofibromas pueden convertirse en malignos, generando así una enfermedad potencialmente fatal.
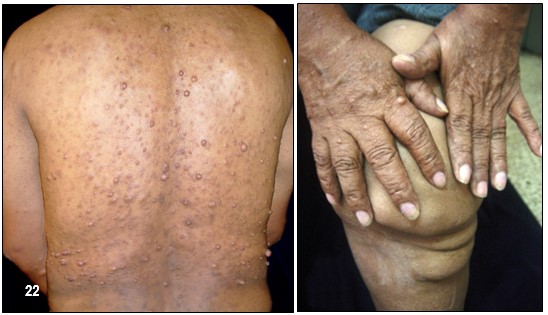
Los neurofibromas están formados de diferentes tipos de células; unas de ellas son las Schwann. Tan solo una de estas células, en estado anormal, no es suficiente para causar el desarrollo de un neurofibroma. Aquella debe agruparse con otras, también anormales, en las cuales el gen NF1 presente errores.
Un neurofibroma se desarrolla cuando ambas copias alteradas del gen NF-1 en una célula Schwann, entra en contacto con un mastocito parcial o totalmente mutado.
La escoliosis, o curvatura de la columna vertebral, pueden afectar la apariencia cuando es grave. Crecimientos pueden ocurrir alrededor del pezón (neurofibromasperiareolar), que puede ser muy molesto. Rara vez, un crecimiento excesivo de la piel o el hueso causa el agrandamiento de un brazo o una pierna, viéndose más a menudo en el síndrome de Proteo.
En algunos pacientes el tamaño o el número de neurofibromas aumenta durante la pubertad y el embarazo, lo que refleja un posible efecto hormonal. No existen evidencias de que ciertos alimentos el ejercicio físico afectan el crecimiento de los neurofibromas.
Existe la creencia científica de poder eliminar la aparición de neurofibromas al prevenir cualquier contacto de las células Schwann con los mastocitos
Una persona sana puede presentar la aparición de un neurofibroma. Ocurre esporádicamente y no indica la presencia de NF. La mayoría pero no la totalidad de los afectados los desarrollan.
Los neurofibromas no son crecimientos cancerosos puesto que no se esparcen ni se riegan por todo el cuerpo pues están fijamente localizados. La mayoría de estos tumores, aún los muy grandes, son benignos y permanecen siempre de esta manera. Por lo tanto, no es necesario llevar un registro pormenorizado de cada fibroma.
Sin embargo, en casos muy raros y aislados se vuelven malignos y se convierten en fatales. Se denomina a esta situación una transformación maligna. Aunque un neurofibrosarcoma (NFS), es raro dentro de la población en general, ocurre con excesiva frecuencia, 50%, entre pacientes con NF-1. Estos tumores son altamente agresivos e invasores, luego un tratamiento rápido es esencial para lograr una alta expectativa de vida. Se presume la presencia de un NFS, hasta no probar lo contrario, cuando las manifestaciones son una masa en continuo crecimiento, dolor localizado en alguna parte del cuerpo o algún factor neurológico.
El riesgo de ocurrencia es de 10-10,000 veces mayor que la población en general. Así mismo, los resultados de las encuestas indican que la edad de los pacientes afectados está por debajo de los 30 años.
Contadas excepciones, un fibroma nunca aparece en el lugar en donde hay una mancha café con leche. Se manifiestan en el cuerpo en forma externa, interna o de ambas maneras, creciendo en cualquier punto a lo largo del sistema nervioso central o periférico.
Existen cuatro tipos de neurofibromas: cutáneos, subcutáneos, difusos o nodulares y plexiformes.
Cutáneos: Los hay de tres tipos: planos, pedunculados (con tallo), y sésiles (inmóviles, sin tallo y adheridos a su propia raíz).
Un neurofibroma cutáneo brota en la piel como un diminuto punto rojo el cual crece hasta asemejarse a una picadura de insecto, de color rojo fuerte y con un punto en la mitad. Al cabo del tiempo, pasa a ser rosado y se convierte en una verruga, de forma ovalada o irregular, con consistencia blanda o semi elástica, suave al tacto y se desplaza al compás de la piel. Al cortarlo es brillante.
Bajo un microscopio se observa una masa de capa serosa compuesta por células del tipo fibroblastosas, fusiformes de citoplasma eosinófilo con núcleos alargados y con producción de colágeno. Hasta la fecha no se ha llevado a cabo un análisis bioquímico de un neurofibroma. Por tal razón se desconoce su composición química. Las células fibroblastos producen la fibra.
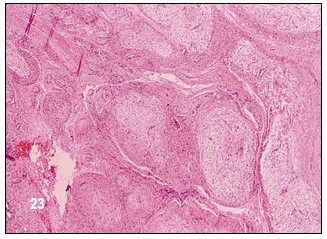
Los neurofibromas de la NF-1 y de la NF-2 son histológicamente distintos. Según estudios microscópicos realizados a su estructura, tanto su tejido como sus cambios genéticos son diferentes.
Hacen su aparición durante la niñez pero regularmente tienden a brotar y a crecer en épocas como la pubertad y el embarazo en las cuales el organismo segrega mayor cantidad de indistinta clase de hormonas tales como el estrógeno y la progesterona. Aun no es posible predecir la cantidad que llegue a aparecer, desde algunos cuantos hasta cientos. Generalmente no causan dolor y los afectados presentan, o ninguno o muy pocos efectos o problemas cosméticos; unas pocas MCL y/o unos pequeños neurofibromas cutáneos poco o nada visibles o fácilmente cubiertos con ropa. Unos pacientes permanecen estables durante su vida adulta mientras en otros se les desarrollan más. No existe evidencia que el ejercicio, la dieta o las vitaminas afecten o causen su crecimiento. Sin embargo, los neurofibromas pueden producir efectos cosméticos y llegar a causar desfiguración.
Los cutáneos pueden desarrollarse en la cara como también en los brazos y en las piernas y en casos alarmantes llegar a aparecer cientos y cientos y en tal eventualidad, una limpieza corporal total es muy difícil y el cepillado de algunas de sus partes se vuelve muy doloroso.
En casos extraños, algunos pueden llegar a crecer hasta el punto de causar el alargamiento de una extremidad y otros a causar malestar tales como aquellos creciendo junto a un pezón. (neurofibromas peri areolares). En casos reportados por la ciencia médica, algunos pacientes con peso corporal de 48 kg, desarrollaron un neurofibroma de 70 kg.; se sometieron a una extraordinariamente delicada intervención quirúrgica para extirparlo y lo lograron. El neurofibroma era benigno y en la actualidad el paciente goza de excelente salud.
-Subcutáneos: Los neurofibromas subcutáneos crecen apenas debajo de la piel y rara vez se desarrollan antes de la adolescencia. En algunas ocasiones duelen y causan rasquiña o prurito. En estados severos comprometen el sistema neurológico.
-Nodulares: Este tipo de neurofibromas, algunos de ellos congénitos, involucran las raíces, el plexo nervioso y la piel a lo largo de toda su extensión. Al crecer pueden llegar a comprometer órganos viscerales.
Otro de los nombres utilizados por el cuerpo médico para la neurofibromatosis tipo 1, es la NF periférica. Sin embargo, los tumores, afortunadamente poco comunes, pueden involucrar el sistema nervioso central. Entre ellos se encuentran los astrocitomas, ependimomas, meningiomas y las metástasis intra medulares.
Investigadores alemanes descubrieron recientemente la presencia de fibras nerviosas en diferentes tipos de tumores. Sus estudios concluyeron que dichas fibras se relacionan exclusivamente con el tumor mas no con el órgano afectado. Por lo tanto, dichos nervios tumorales están involucrados con el sistema nervioso autónomo. Inicialmente se cree que el estrés y otras características sicológicas pueden influenciar la aparición de tumores en el organismo. Aun se desconoce la función conectora de estas fibras nerviosas pero que dan lugar a un tipo de neuro transmisión.
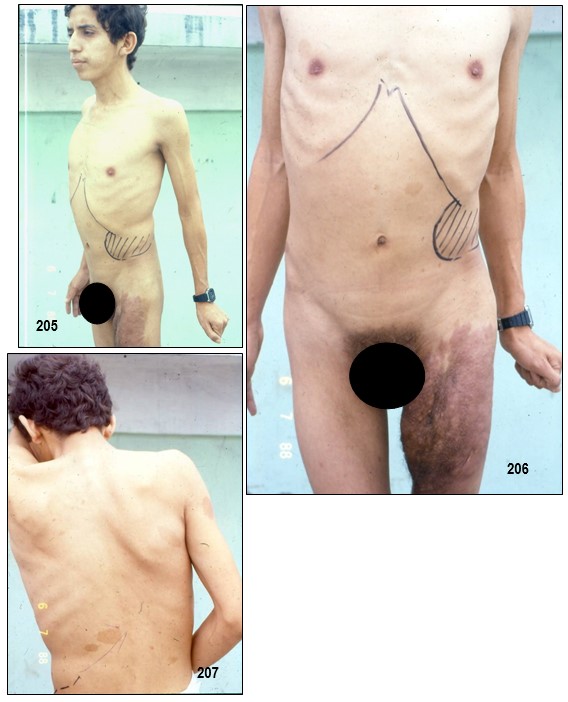
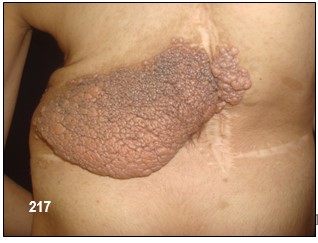
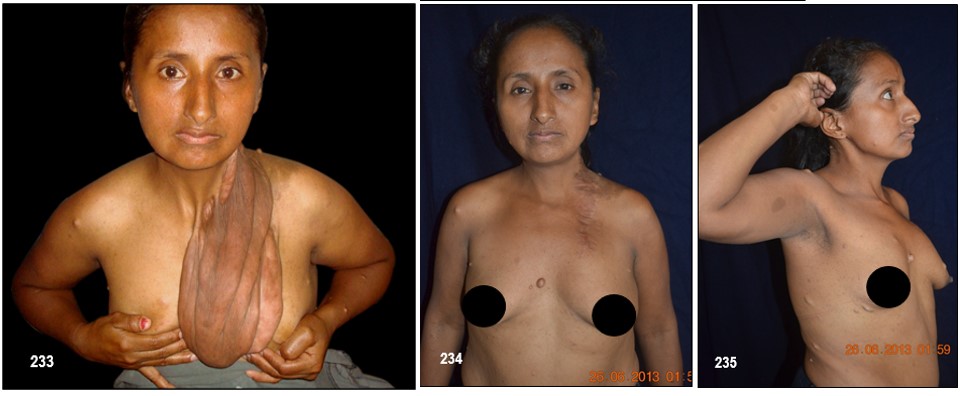
Neurofibromas plexiformes.- Un plexo es una compleja red de nervios o de vasos sanguíneos. Reciben el nombre de neurofibromas plexiformes los que afectan las múltiples ramas de un nervio más o menos grande y se deben a un mayor crecimiento de células Schwann. Son una característica específica de la NF-1.
Generalmente se desarrollan durante la niñez y su crecimiento está delimitado. Durante el primer año de vida del paciente, este tipo de neurofibroma puede asemejarse a un tejido suave alargado -difuso – o a un parche de hiper pigmentación cutánea.
Su presencia pasa desapercibida hasta cuando crecen. Pueden convertirse en masas gigantescas, con formas aberrantes y llegar a comprometer tejidos sanos circundantes.
Comúnmente, son tumores congénitos difusos pero a veces son nodulares llegando a desarrollar múltiples tumores discretos.
Muchos de ellos crecen internamente, en el tronco y en las extremidades, asintomáticamente como para pasar desapercibidos durante exámenes de control, pero a veces se presentan alrededor de un ojo causando un alargamiento en el párpado superior el cual tiende a crecer durante el transcurso de los años. Así mismo, pueden afectar a un lado de la cara.
Los neurofibromas se pueden desarrollar entre las vértebras comprimiendo la columna vertebral o algún órgano adyacente y son muy difíciles de manejar para su extirpación. Se les denomina neurofibromas de campana hueca.
Entre los órganos susceptibles de ser comprimidos o desplazados figuran la médula, la pleura, el saco dural, etc. Algunas veces se puede presentir, desde un comienzo, cuando por ejemplo el caminado se vuelve como tipo soldadito de plomo, se siente asfixias, no se controlan los esfínteres, etc. En relación con las asfixias, en algunas ocasiones suelen presentarse en la región del foramen causando complicaciones respiratorias.
Los plexiformes también pueden aparecer en la región del mesenterio y en tal caso, por lo general, quienes los padecen, sufren de dolores abdominales y vómitos.
Los afectados con plexiformes en el párpado suelen también padecer de displasia esfenoidal en mismo lado del ojo afectado. Aunque esta displasia es asintomática, puede asociarse con una hernia a través del defecto óseo. Así mismo, pueden llegar a comprometer el canal auditivo externo y, en consecuencia, afectar la audición.
Un plexiforme puede venir acompañado de una hiperpigmentación y/o de un crecimiento excesivo de pelo (hipertricosis), tal como se presenta en la foto 9; adviértase pelo debajo de la tumoración. Pueden causar erosión ósea y dolor.
Ellos son muy difíciles de suprimir completamente pues tienden a reaparecer. Por lo tanto, su erradicación debe ser previamente y fielmente estudiada. Se caracterizan por su altísima tendencia a sangrar y por la inhabilidad de lograr su erradicación total en caso de cirugía.
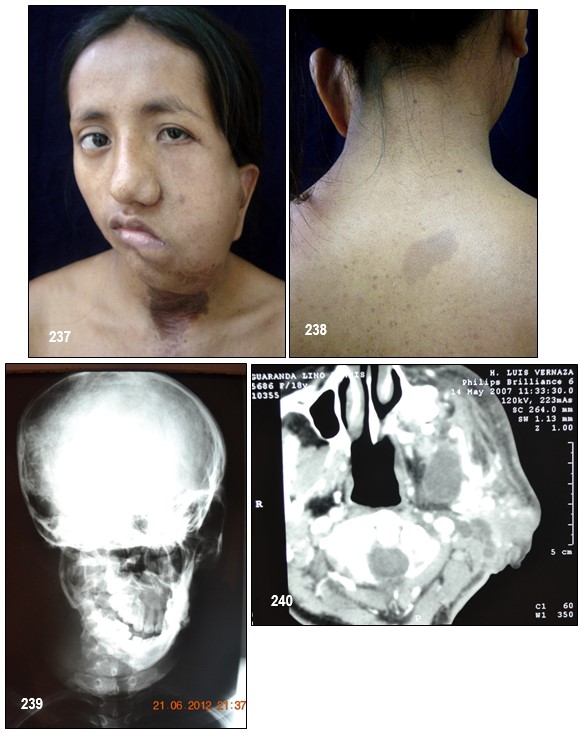
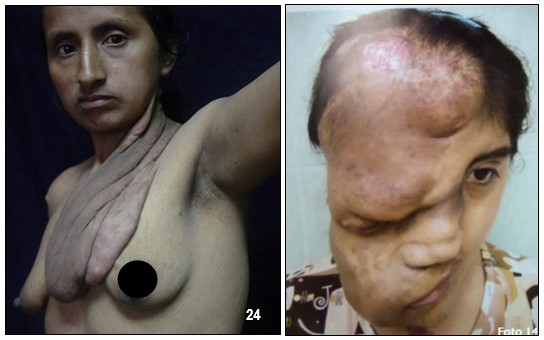
Por lo general, un neurofibroma no se maligniza pero de llegar a ocurrir, se presentaría con mayor seguridad en un plexiforme (hecho que motivo a la extirpación del mismo a la paciente de la las imagen 233). Un crecimiento rápido y súbito o síntomas de dolor o flogosis inexplicada donde antes no lo hubo deben evaluarse inmediatamente con el fin de descartar un sangrado o una transformación maligna pues este tipo de tumores malignos periféricos son altamente agresivos y metastásicos debido a tener propensión a crecer y afectar tejidos adyacentes normales. En tal caso, se deben hacer varias biopsias de diferentes regiones del tumor plexiforme para descartar cualquier tipo de cambios malignos.
La malignidad neoplásica ocurre en el 5% de los casos y se combate mediante una combinación de cirugía, radiación y quimioterapia. Se puede llegar a presentar, con estrecha relación a un neurofibroma plexiforme, un tumor muy extraño llamado Tritón.
Se hace notar que los tumores plexiformes pueden llegar a doler en caso de ser traumatizados o golpeados, pero este síntoma es totalmente diferente al maligno el cual ocurre sin evidencia externa alguna. Es de consuelo saber que en personas normales una malignidad ocurre en el 25% de los casos.
Hasta la fecha no se ha observado ninguna característica clínica específica de la NF-1 asociada con la presencia de neurofibromas plexiformes.
Efélides en axila o ingle (signo de Crowe).- Generalmente, esta manifestación de manchas lentiginosas en la axila no se presenta en personas con NF-1 (80% a los 5 años de edad). Sin embargo, la presencia de más de tres es una evidencia muy fuerte de la existencia de la alteración genética y es el signo más confirmativo después de las MCL.
Estas manchas, plenamente comprobadas, están esencialmente restringidas a esta clase de zonas en donde hay doble contacto de piel, llamadas intertrigas y probablemente el origen de estas pecas tenga un mecanismo diferente al de las MCL. Las MCL no son comunes al nacer. Aparecen entre los tres y los cinco años de edad, aunque las he observado en edades más tempranas. Por lo general, su tamaño no pasa de 5 mm. Se encuentran primordialmente en axilas, ingles, sobre los párpados superiores y alrededor del cuello, algunos pacientes las tienen diseminadas en cara alrededor de los labios, debajo de la quijada, tronco y parte proximal de las extremidades, en mujeres pueden encontrarse debajo de las mamas.
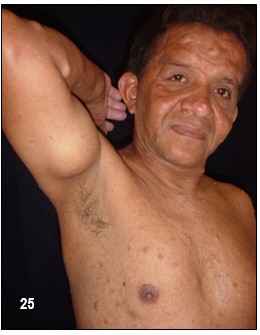
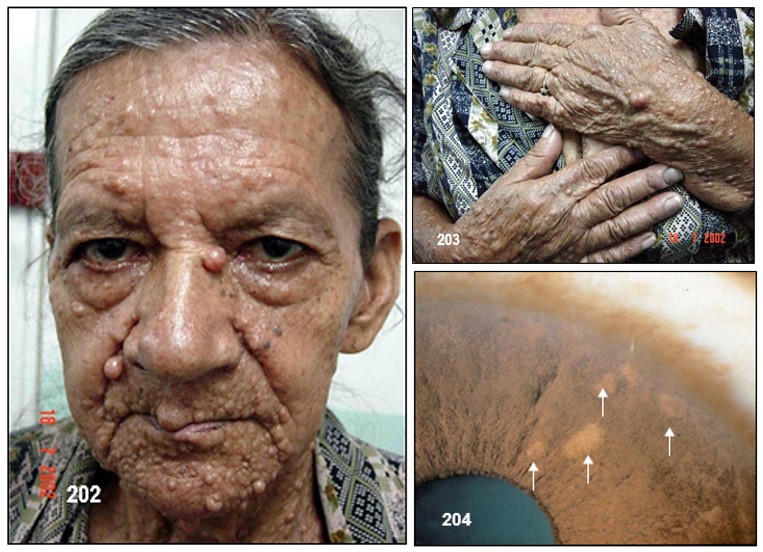
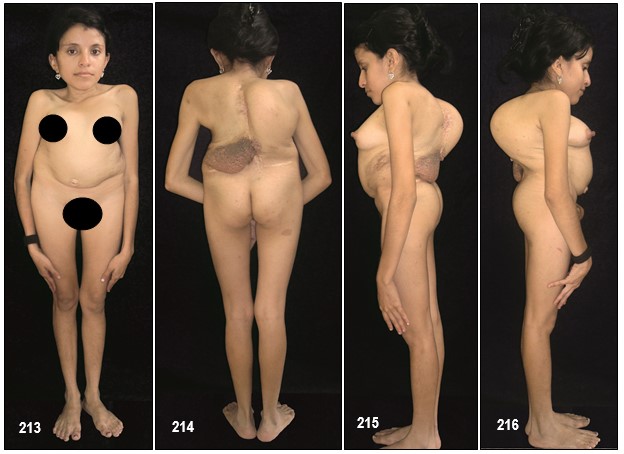
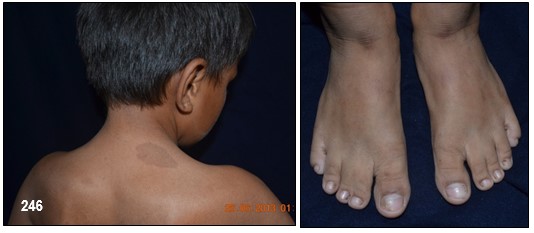
Imagen 25 con fines Didácticos.- Enfermedad de Von Recklinghausen. NF-1. Neurofibromas cutáneos y subcutáneos; lentigos o pecas (efelidoides) a manera de máculas hiperpigmentadas menores de 2mm distribuidas difusamente y que comprometen las axilares (signo de Crowe). La presencia de pigmentación en forma de pecas en las axilas, ingles y el periné se considera patognomónica de la enfermedad. Nótese varios neurofibromas plexiformes, a nivel del brazo uno subcutáneo de gran tamaño y otros más pequeños a la altura del antebrazo (Fuente: Tama Viteri F. Atlas y Texto en Color de Imágenes Clínicas Segunda Edición 2011. Senefelder. Universidad de Guayaquil).
Así mismo se presentan variaciones debidas a la etnia o raza de la persona. Las pecas comunes son poco frecuentes en negros y asiáticos.
En los pacientes pediátricos que presento he podido observar con asombro la presencia de acantosis nigricans en grandes pliegues, sobre todo subglúteo; si consideramos a esta manifestación como expresión de una resistencia a la insulina se procedió a valorar esta alteración metabólica, sin encontrar su relación, invitándonos a pensar más bien en una manifestación seudoacantosica muy común en esta etnia.
Defectos congénitos de la médula.- La variedad de defectos óseos observados en NF1 suelen ser evidentes al nacer. La mayoría son poco comunes. Los defectos pueden ocurrir en cualquier hueso, pero se observan con mayor frecuencia en el cráneo, la columna y las extremidades. Ellos incluyen:
- -Ausencia congénita de la pared orbital. Su ausencia puede provocar una ligera protuberancia en la piel alrededor del ojo.
- -Arqueo de los huesos de la pierna debajo de la rodilla (tibia o el peroné). Estos huesos pueden ser más delgados de lo normal. Si se produce una fractura, la curación puede ser lenta o incompleta.
- -Cicatrización incompleta, llamado pseudoartrosis, también puede afectar a los huesos del antebrazo (radio y cúbito), pero esto ocurre muy raramente. Este es un problema difícil, que requiere la supervisión de un cirujano ortopédico.
Nódulos o nevus en el ojo.- Los tumores o nevus en el iris del ojo se llaman nevos, hamartomas o nódulos Lisch en honor del oftalmólogo austriaco Karl Lisch.
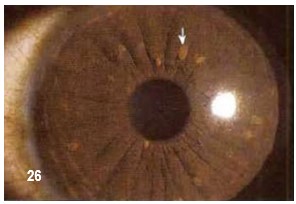
Los nevus en el iris, se trata de hamartomas que aparecen sobre el iris y se puede distinguir de las pecas del iris (comúnmente visto en personas sin NF) por un procedimiento simple y sin dolor llamado un examen con lámpara de hendidura, que se lleva a cabo típicamente por un oftalmólogo. Aumentan en número y tamaño con la edad. Se presentan en un 10% a los 10 años y hasta 90% adultos. Raramente presentan una sintomatología significativa. Los nevus del iris no causan problemas médicos y no afecta la visión. La presencia de nevus del iris en ocasiones puede ser útil para confirmar el diagnóstico de NF.
- -Anormalidades coroideas: Del 50 hasta el 100% según las series presentan anormalidades coroideas durante su evolución, pero son difíciles de identificar. Precisan de verde indiocianina con luz infrarroja y oftalmoscopio láser con scanner para objetivarlas.
- -Ptosis palpebral: aparece aislada (9% de casos) o asociada a un neurofibroma.
- -Glaucoma congénito: 0.5% de casos.
En el evento de no existir familiares con neurofibromatosis, ambos padres deben examinarse el iris de sus ojos mediante un examen bio-microscopico binocular por medio de una lámpara de hendidura para verificar la posible presencia de nódulos Lisch en ellos; es la característica más común en la NF-1.
La presencia de más de uno de ellos confirma la NF-1. Estos nódulos no afectan la visión ni producen problemas médicos. Son masas de pigmentos. Al parecer, este rasgo es un síntoma exclusivo de este trastorno genético y aparecen durante la pubertad.
Los nódulos Lisch están relacionados con la edad mas no con el número de manchas café con leche en la piel, ni con el número de neurofibromas o ni con la severidad del trastorno genético.
Como se puede apreciar en la imagen en el texto, estos nódulos tienen forma de domo y su color oscila entre claro amarilloso hasta café oscuro, dependiendo de la etnia, desde la raza caucásica hasta la negra. Así mismo, el color del iris del ojo no tiene relación alguna ni con la presencia ni con la cantidad de ellos. Su diámetro es menor a los 2 mm. Su contorno es suave y son translucidos.
Los nevus del iris no interfieren con el uso de lentes de contacto de ninguna clase ya sean blandos, duros, permanentes, de uso prolongado, etc.
Dos o más de estos nódulos de Lisch son un criterio invaluable para poder diagnosticar la NF-1 en casos difíciles o incompletos. Por lo general, el 50% de los pacientes adultos con NF-1 los tienen y el 100% de ellos los tendrán al entrar en la sexta década de sus vidas.
Las tecnologías recientemente desarrolladas en la Universidad de Osaka, Japón, han arrojado como resultado un gran compromiso del coroideo en la NF-1. Estas investigaciones sugieren el complementar el diagnostico de este cambio genético ante la presencia de parches brillantes en la región coroidal. Dichos parches solo pueden ser observados mediante el uso de equipos láser oftalmoscópicos emisores de luz infrarroja puesto que esta penetra desde el epitelio del pigmento retinal hasta el coroideo a diferencia de los resultados obtenidos mediante el uso de otra clase de equipos y procedimientos oftalmológicos. Estos investigadores atribuyen estas regiones con parches a la presencia de tejido retráctil en el coroideo el cual es una de las partes más comúnmente afectadas por la NF-1.
Otras manifestaciones oculares esta el glaucoma, y el hipertelorismo.
DEFORMACIONES ÓSEAS- Desfiguración.– La NF-1 puede producir desfiguración en un sin número de maneras. La deformación puede ocurrir casi en cualquier hueso pero por lo general se presentan en el cráneo y huesos de la cara, así como en las extremidades, y a nivel de la columna vertebral, por lo cual se recomienda un detenido examen óseo.
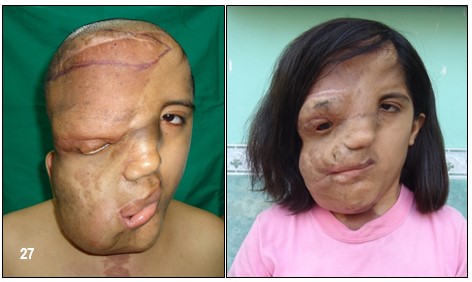
Los neurofibromas cutáneos pueden desarrollarse en la cara (Imágenes con fines Didácticos) o en las áreas expuestas de los brazos o las piernas. Los neurofibromas plexiformes más grandes y profundos pueden crecer alrededor del ojo, el párpado, o afectar el crecimiento de un lado de la cara. La escoliosis, o curvatura de la columna vertebral, pueden afectar la apariencia cuando es grave. Crecimientos pueden ocurrir alrededor del pezón (neurofibromasperiareolar), que puede ser muy molesto. Rara vez, un crecimiento excesivo de la piel o el hueso causa el agrandamiento de un brazo o una pierna, viéndose más a menudo en el síndrome de Proteo.
En algunos pacientes el tamaño o el número de neurofibromas aumenta durante la pubertad y el embarazo, lo que refleja un posible efecto hormonal. No existen evidencias de que ciertos alimentos el ejercicio físico afectan el crecimiento de los neurofibromas.
El temor a la desfiguración a menudo son las principales preocupaciones para los afectados con NF-1. Algunos pacientes encuentran que las manchas de color café con leche o un número mínimo de neurofibromas cutáneos son difíciles de vivir, mientras que otros son capaces de soportar una participación más grave. Si se desea la cirugía, principalmente para mejorar la apariencia, un cirujano plástico puede ser consultado para determinar si un tumor particular puede ser retirado. Neurofibromas plexiformes alrededor del ojo a menudo son manejados en forma conjunta por el cirujano oftalmólogo y un cirujano plástico.
Generalmente no ocurren pues son síntomas relativamente extraños. De llegar a presentarse deformaciones óseas, estas se manifiestan desde el nacimiento. No aparecen de un día para otro. Un niño de 5 años con exclusivamente MCL, casi con absoluta seguridad se ha librado de tener al menos alguna de las complicaciones graves de la NF-1.
La escoliosis se manifiesta en un 10% – 20% de los casos. Por sí sola no es una característica propia para diagnosticar la NF-1.
Puede aparecer durante la niñez, como es el caso de unos de los pacientes pediátricos presentados.
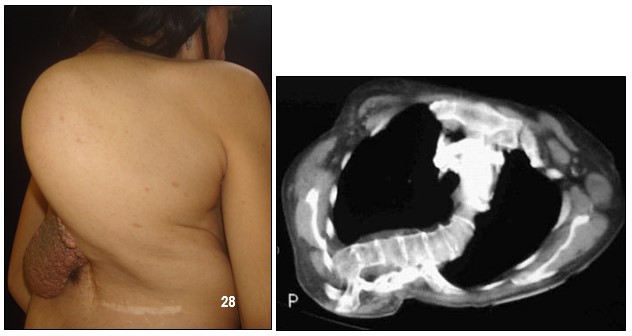
Los síntomas de la escoliosis son dolores en la espalda o en las extremidades inferiores persistentes y progresivas sin causa aparente, desbalance al caminar, un déficit neurológico y una deformidad anatómica creciente. Esta enfermedad progresa con el tiempo y genera problemas cosméticos y psicológicos y puede llegar a comprometer el entorno cardiopulmonar.
En la mayoría de los casos se presenta en forma muy suave pero, en caso grave, afectan la apariencia del paciente y de manera especial a las mujeres jóvenes.
En el evento de padecer de escoliosis lumbar, dolor de espaldas o insensibilidad en los pies puede ser debido a unas malformaciones arteriovenosas epidurales lumbares. Estas son poco comunes y por lo general están localizadas en la región cervical. Los tumores en la columna vertebral pueden llegar a producir parálisis.
La displasia puede producir un arqueo congénito de la tibia o del peroné, por lo general en solo uno de ellos, Las cortezas de estos huesos se tornan más delgadas de lo normal por lo cual se vuelven más propensos a fracturarse y su curación puede ser lenta o incompleta la cual recibe el nombre de seudo-artrosis. Se presenta en un 1% – 2% de los afectados y con mayor incidencia en el sexo masculino. Pocas veces se llega a casos extremos de amputación.
No es de ayuda alguna el uso de un yeso. Por lo general, se necesita una cirugía. En casos extremadamente raros, menos del 1% de los pacientes requieren de una amputación. De igual manera se pueden afectar los huesos del antebrazo, el radio o el cubito, pero esto se presenta muy rara vez. Todas estas son condiciones extremadamente delicadas y requieren de la ayuda de un ortopedista pues la anomalía puede incidir en un crecimiento corporal defectuoso.
A veces se presenta una deformidad en la pared ósea detrás de la órbita del ojo. Algunos recién nacidos tienen una anomalía en los huesos esfenoidales (displasia del ala mayor). Esta anormalidad se detecta mediante exámenes de rayos X o con tomografías. No es necesario efectuar procedimiento alguno cuando se descubre la ausencia de este hueso. Esta deficiencia puede producir un abultamiento o exoftalmia alrededor del ojo o una caída de éste. Es de vital importancia un pronto y eficaz tratamiento de este síntoma con el fin de evitar y prevenir cualquier tipo de fractura.
Se pueden presentar anomalías vasculares a lo largo de la columna. Estas deben ser sometidas a consideración en un diagnostico diferencial de cualquier tipo de masa epidural relacionada con esta modificación genética. Se ha descrito también pseudoartrosis.
Dificultades en el aprendizaje.- Existen dos causales para la aparición de dificultades o problemas en el aprendizaje: físicas y emocionales. Las físicas aparecen en aquellos casos en que se compromete el sistema nervioso central mediante interferencias en la recepción y en el procesamiento de datos del cerebro. Dichas deficiencias se originan tempranamente en el embarazo, en la etapa de formación del sistema nervioso.
Entre los problemas serios de la NF-1, este es el más común. Ocurre en un promedio de seis veces mayor en personas afectadas que al de la población en general y en aquellas, se presenta en el 50% de los casos. Su grado varía entre cada afectado. Supuestamente, este inconveniente proviene de anomalías cerebrales: tumores e hiperintensidad.
Existen razones de peso para sospechar que la NF-1 conlleva algún tipo de deterioro en el desarrollo intelectual. La dificultad en el aprendizaje se puede catalogar como un corto circuito en uno o en varios de los canales cerebrales.
Estudios realizados tanto en Suecia como en otros lugares han arrojados resultados evaluando que, por lo general, las personas afectadas con NF-1 presentan retardo mental ligero. Estos problemas en el aprendizaje no se complican con el paso del tiempo. No son progresivos, pero no desaparecen al llegar a la edad adulta. No existe patrón específico sobre las dificultades en el aprendizaje relacionadas con la NF-1. Investigaciones recientes canadienses relacionan las diferentes dificultades en el aprendizaje con un extremado bajo peso corporal en el momento del nacimiento.
Todo ser humano tiene dos copias de cada gen en su cuerpo. Quien presenta una afección por NF-1, tiene una de las copias del gen NF-1 deteriorada, errada o mutada en cada una de las células de su cuerpo; es decir contiene una copia normal y otra anormal.
El tener una de estas últimas en la región cerebral, predispone a la existencia de dificultades en el aprendizaje.
Un grupo de científicos norteamericanos, encabezados por el Dr. Alcino Silva, encontraron en ratones de laboratorio con una mutación de NF-1, unas señales inhibidoras del cerebro inusualmente fuertes e hiperactivas. Se cree que durante el proceso de aprendizaje, las señales estimulantes cerebrales efectúan cambios en la estructura del cerebro necesaria para la memoria. Es probable que tanto en los ratones como en las personas con NF-1, tales señales no son lo suficientemente fuertes para contrarrestar la anormalmente alta inhibición causada por la mutación de NF-1 y, como consecuencia, se pierde gran cantidad de información antes de llegar a conservarse. Dichos animales presentaron deficiencias de aprendizaje con una sorprendente similitud a las de los humanos. La antedicha mutación de NF-1 produce un aprendizaje espacial deficiente.
Una de los posibles causales de los problemas de aprendizaje radica en contener el cerebro una insuficiente cantidad de la proteína neurofibromina. Sin embargo, en estudios realizados por el Dr. Silva, en la ciudad de Los Ángeles, EEUU, sobre tales roedores, dichos problemas se pueden eliminar mediante la aplicación de drogas bloqueadoras. Según tales estudios, dichos fármacos arrojaron infinidad de efectos secundarios en humanos.
Entre las dificultades se encuentran los problemas de atención, las deficiencias de memoria, las deficiencias del lenguaje, los problemas de organización y las disfunciones motrices gruesas y finas. Tales deficiencias son el efecto de una organización cerebral diferente. Se clasifican en tres grupos definidos: global, habla, y retardos motrices. Cualquiera de ellas debe de ser reconfirmada por el neurólogo en varias sesiones con el paciente.
Los problemas en el aprendizaje, aunque no son una enfermedad, son tal vez el síntoma más devastador en la vida de los afectados pues contribuyen a una muy baja imagen personal, deficientes experiencias educativas y reducidas oportunidades para lograr una satisfacción personal o para obtener trabajos bien remunerados. Por tal motivo, estas deficiencias de aprendizaje deben ser atacadas desde temprana edad, mediante tratamientos con valoraciones psicológicas individuales, y total apoyo familiar, con el fin de prevenir en el futuro del paciente problemas personales, sociales y económicos tales como baja autoestima, formación académica irregular, deserción escolar, delincuencia juvenil y formal, drogadicción, desempleo, dificultades matrimoniales y familiares.
Los síntomas se empiezan a manifestar en los primeros años de edad escolar. Por lo general, con dificultad con las destrezas visuales o espaciales, también en el habla y el lenguaje, a veces en la lectura o con las matemáticas o cualquiera de estas combinaciones. Comúnmente, la dislexia se debe a una anomalía genética o a un daño cerebral. También se manifiesta en una falta de atención la cual puede subsanarse aplicando medicamentos estimulantes adecuados. Adicionalmente, el promedio en la coordinación fina motriz decrece.
En algunas circunstancias, las agudezas visuales o auditivas pueden ser perfectas, sin embargo, la manera como el niño percibe u oye su entorno puede fallar. El niño lee “ola” en lugar de “alo”, confunde el “3” con la letra “E”, etc. Repite líneas al leer, confunde la izquierda con la derecha, se salta palabras. No calcula distancias ni la profundidad, se tropieza o tumba objetos, confunde sonidos similares, tienen poca percepción de expresiones faciales, tono de voz, tiene pocos amigos, poca memoria, mala motricidad, mala percepción espacial.
Por lo tanto, los profesores, médicos y padres deben ayudar a detectar desde temprana edad la presencia de estos síntomas, darles una especial atención y trabajar en conjunto para controlarlos antes de llegar a presentarse una falla escolar y el niño comience a perder confianza en sí mismo. Se debe alabar sus cualidades y logros en lugar de recalcar sus fallas. Estos pacientes deben ser observados y evaluados por un sicólogo o un neurólogo pedíatra.
Es muy importante tener siempre bien en cuenta que los niños con problemas de aprendizaje son iguales a cualquier otro niño. La diferencia radica en el modo de aprendizaje. A estos niños se les acusa de no hacer el esfuerzo suficiente para poner atención cuando en realidad lo están haciendo mejor que sus compañeros de clase.
Investigaciones recientes mediante el uso de imágenes de resonancia magnética, han demostrado, en infantes con NF-1, la presencia de objetos brillantes no identificados, Obnis, en la región talámica, está estrechamente relacionada con coeficientes intelectuales menores, baja memoria, motricidad, fácil distracción, bajos dominio de atención; entre mayor cantidad de Obnis, menor el coeficiente intelectual, IQ. Las biopsias practicadas a estas lesiones demuestran un mayor contenido de agua en ellas sin llegar a presentarse displasia. Se les considera un tipo de hamartoma benigno. La presencia de estas imágenes en otras partes del cerebro no tienen ningún impacto en el desarrollo neurofisiológico. Por lo general, estos objetos desaparecen con la edad.
El retardo mental de grado más severo correspondiente a un nivel escolar especial no es típico de este trastorno genético. No obstante de todo lo anterior, en caso de presentar un paciente dificultades en el aprendizaje, estas pueden no deberse a la NF-1.
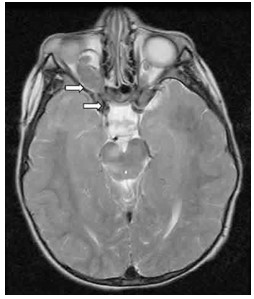
Gliomas o tumores ópticos.- El nervio óptico controla la vista. Un glioma óptico es un tumor en cualquier lugar de este nervio pero por lo general se desarrollan en la región junto al hipotálamo cerebral. Esta clase de tumores es poco frecuente, tan solo en un 15%, y aparece durante la niñez, de 4 a 6 años, y es fácilmente identificado por mala o decreciente agudeza visual o por un abultamiento en el ojo llegando a afectar la secreción de la hormona pituitaria.
En asocio con un glioma óptico pueden ocurrir sutiles defectos de campo periféricos, dificultad en la discriminación de los colores, palidez del nervio óptico, distorsión en la mirada binocular, glaucoma (en casos muy aislados), cefaleas o un desplazamiento del ojo (proptosis), estrabismo, sin llegar a causar problemas de agudeza visual.
Sin embargo, tanto los gliomas ópticos como los astrocitomas son las clases más comunes de tumores del sistema nervioso central en personas con NF-1. Se clasifican en dos clases. La variedad más común de gliomas benignos es aquella asintomática; se detectan tan solo al llevar a cabo exámenes de resonancia magnética. Los otros se comportan de manera agresiva y pueden comprometer la agudeza visual y causar estados enfermizos.
Un astrocitoma es una clase de tumor de bajo perfil o glioma, y se da como resultado de anomalías genéticas. Los estados tumorales o tumorgénesis se presentan cuando existe una falta parcial o total del gen supresor.
Es recomendable una resección parcial de un astrocitoma cuando se localiza cerca de áreas importantes cerebrales.
Los gliomas de la retina son extraños y usualmente están relacionados con la NF. Estos tumores blanquecinos por lo general contienen calcio y se asemejan a un pequeño retinoblastoma. Probablemente son malformaciones o tumores benignos llamados hamartomas y tienen un potencial de crecimiento limitado. Un neurofibroma es una clase de hamartoma. Una vez detectados, las anormalidades visuales poco progresan. Se aconseja tratamiento médico y quirúrgico (quimioterapia / radioterapia) para aquellos casos aislados que lleguen a progresar. En tal caso, el paciente debe estar previamente advertido sobre los efectos secundarios de las altas dosis radioactivas acerca de una posible aparición y desarrollo de neoplasmas malignos. En algunos casos no tratados a tiempo estos tumores en el nervio óptico pueden llegar a producir ceguera pero por lo general no afectan a ambos ojos.
Su presencia no está relacionada con ningún otro factor de riesgo tales como ocular o pérdida visual, óseo, neurológico o debido a la anamnesis del paciente.
Las investigaciones realzadas sobre gliomas del tallo cerebral han encontrado una suma cuantitativa de absorción de talio. No existe relación alguna entre la cantidad absorbida por el organismo y el tamaño del tumor y se desconoce el origen de dicha absorción.
Algunas evidencias señalan a las razas Afro-americanas de tener un menor riesgo de padecer glioma del nervio óptico en contraste con la raza blanca junto con los hispanos.
La localización del glioma es el factor más importante para poder determinar la naturaleza de los síntomas y de las señales. Puesto que por lo general, estas lesiones, la gran mayoría benignas, crecen lentamente, los ataques pueden considerarse como el síntoma clínico y pueden sucederse mucho tiempo antes del diagnóstico. Por lo general, un ataque demuestra la presencia de un tumor en los lóbulos temporales y frontales.
La mayoría de los gliomas son invasivos; pocas veces son lesiones puntuales. A pesar de esto, a menudo se pueden controlar y curar mediante tratamiento con resección radical. En caso de presentarse ataques, un electroencefalograma puede utilizarse como diagnóstico al emitir ondas y actividad epileptiforme.
Un tumor puede conllevar a presión intracraneal, deficiencias neurológicas progresivas, cambios sensoriales, dificultad en el habla y en el lenguaje.
Hipertensión arterial.- La Hipertensión arterial (HA) es otro de los síntomas de la NF-1. Es muy probable encontrar hipertensión en niños con NF-1. Se está comprobando una posible relación del gen de la NF-1 con las enfermedades cardiovasculares.
La HTA es un hallazgo frecuente en pacientes con NF-1 (1,5–2,5%) Puede considerarse esencial, pero lesiones vasculares que provoquen HTA renovascular son más frecuentes en pacientes con NF1.
En casos de coexistencia de HTA y NF1, se ha detectado un feocromocitoma en el 20–65% de los casos.
Tener en cuenta la posible existencia de un feocromocitoma, presente en el 0,1–5,7% de los de los pacientes con NF1. De los cuales: 84,2% unilateral; 9,6% bilateral y 6,1% ectópicos. La edad media de diagnóstico son los 42 años.
Manejo de la HTA: monitorización periódica de la HTA y screening de estenosis de arteria renal y feocromocitoma.
Ante cualquier masa suprarrenal hallada de forma incidental en un paciente con NF1 debe descartarse la presencia de un feocromocitoma.
Importante realizar el diagnóstico preciso de feocromocitoma ante la posibilidad de embarazo, ya que se ha asociado a un aumento de la mortalidad fetal como materna. Importante realizar consejo genético a los padres.
Complicaciones raras de NF-1.- Las complicaciones de la NF-1 se deben a afectación directa de los diversos sistemas por los neurofibromas, a un riesgo incrementado de malignización (la mayoría son fibrosarcomas, pero también otros tumores malignos, inclusive leucemias) y a una serie de asociaciones mal comprendidas.
Las complicaciones se mencionan a continuación ocurrir en 1%, o menos, de los pacientes con NF-1. El anuncio está destinado a informarle de las raras complicaciones posibles en la NF-1, pero hay que destacar que muy pocas personas con NF-1 se experimentan alguno de estos síntomas:
- –Macrocefalia
- -El inicio temprano o tardío de la pubertad.
- -Problemas con el crecimiento (demasiado corto o demasiado alto).
- -El retraso mental.
- -Epilepsia.
- -Escozor cutáneo
- -Los tumores cerebrales (excepto neuroma acústico y glioma óptico).
- -Oclusión vascular cerebral (ictus).
- -Ortopédicos Displasia de alas del esfenoides, escoliosis, arqueamiento congénito o pseudoartrosis, quistes óseos, hipercrecimiento óseo; sindactilia.
- -Endocrinos como talla corta, trastorno neuroendocrino debido a tumores hipotalámicos, pubertad anormal, feocromocitoma.
- -Impacto psicosocial
Los pacientes con NF-1 pueden tener un riesgo algo mayor de ciertos tumores malignos raros que ocurren en el cerebro, los nervios o la médula espinal, pero probablemente tienen el mismo riesgo de cáncer «comunes» (como el de pulmón, mama, estómago, etc.) como en la población general.
Me referiré someramente a algunas de estas complicaciones exóticas en el contexto de la NF-1, compleméntese con el tabla 9..
Macrocefalia.- La macrocefalia por sí sola no es una característica propia para diagnosticar la NF-1. Por lo general, tanto niños como adultos con NF tienen la circunferencia de la cabeza grande lo cual no indica un problema médico de consideración como tampoco significa la presencia de problemas en el aprendizaje. La cabeza crece a un promedio mucho mayor pero en una rata continua y sin modificación. Si este es el caso, no son necesarios ninguna clase de exámenes radiográficos ni de tomografías pero se debe hacer un seguimiento periódico.
A veces, la cabeza sigue creciendo pasada la etapa de la pubertad. Se recomienda medir la circunferencia de la cabeza durante los tres primeros años de vida. En casos extremadamente extraños, se puede producir una hidrocefalia la cual es un problema muy grave y requiere de cirugía.
Pubertad precoz.- Por lo general, la pubertad llega en su edad normal, niñas (8 – 13) – niños (9 – 14). Sin embargo, se puede desarrollar o muy temprano o muy tarde. La pubertad precoz con un crecimiento acelerado ocurre en algunos individuos con NF-1.
La pubertad comienza cuando se desarrollan cambios entre los sistemas de las gónadas y la glándula pituitaria e hipotalámica. Durante este período se incrementa la producción corporal de esteroides de las gónadas y de la glándula suprarrenal.
Los niños y niñas con pubertad precoz tienen en sus organismos mayores cantidades de hormonas como el estradiol y la folicular y las segregan a temprana edad. Entre los síntomas de esta precocidad, figuran: crecimiento de masa corporal, de senos, vello púbico y huesos,
El endocrinólogo debe verificar la aparición de tales características sexuales secundarias con exámenes tales como el de las etapas Tanner. En estos casos, se debe evaluar la posibilidad de una anormalidad en el sistema nervioso central como un neurofibroma, un glioma o una lesión quiasmal interrumpiendo el eje hipotalámico-pituitario del cerebro.
Entre jóvenes afectados con NF-1, la pubertad precoz se presenta más en niños que en niñas y casi siempre asociada a gliomas de la vía óptica con afectación primaria o secundaria del hipotálamo.
Estudios recuentes descubrieron un gen localizado en uno de los pares del cromosoma 19 necesario para dar inicio a la pubertad. Un ejemplar sano es suficiente para comenzarla. Pero dos defectuosos -uno de cada padre- pueden evitar que el cuerpo madure.
Tales investigaciones estiman que un retardo puberal puede deberse en casos de familias en las que se han casado primos pues el gen podría afectar la producción o el procesamiento de una hormona sexual en particular. Así mismo, los factores ambientales, la nutrición, la exposición a diversos agentes químicos e incluso el ejercicio físico también desempeñan un papel en su inicio.
Talla.- La baja estatura por sí sola no es una característica propia para diagnosticar la NF-1 y hasta el momento, la medicina desconoce la relación existente y sus posibles causas.
La estatura de personas con NF puede ser o muy baja o muy alta. Esta condición se presenta en el 25% de pacientes en la etapa de pre adolescencia y en el 43% de adultos en los casos de NF-1 pero no indica un problema médico. A veces se produce una rata de crecimiento anormal: O muy rápida o muy lenta. Un niño con NF-1 es por lo general más bajito que el resto de su familia. Cualquier tratamiento médico o radiológico es por lo general improductivo a menos que la rata de crecimiento cambie repentinamente o preexistan otras manifestaciones diferentes a la NF como por ejemplo, un promedio de baja estatura familiar.
Entre los factores de riesgo para una baja estatura en niños afectados con NF-1, se encuentran las lesiones supraselares las cuales pueden conducir a una deficiencia en la hormona de crecimiento, deformaciones óseas, e insuficiencia nutricional.
Se aconseja efectuar un examen para conocer la secreción de la hormona de crecimiento presente en el organismo. Se estima una deficiencia en los niños afectados independientemente de cualquier daño pituitario o explicación de tipo clínico pero esta falta puede tratarse mediante hormonas de crecimiento y un uso terapéutico de la somatotropina.
Entre las enfermedades que se confunden con la NF, debido a una baja estatura, figuran los síndromes de Noonan y de Watson los cuales no tienen relación alguna con la NF-1.
Otras complicaciones.- Entre otras complicaciones que se presentan, se dan: epilepsia, pruritos o rascado de la piel, oclusión cerebro vascular (bloqueo de los vasos sanguíneos que irrigan el cerebro), descoordinación y anormalidades sensoriales, leucemia mieloide crónica juvenil.
En casos muy aislados, se pueden presentar rabdomiosarcomas y xantogranulomas juveniles. Estos últimos son alteraciones benignas caracterizadas por lesiones cutáneas, o tumores amarillentos, de tejido de granulación con depósitos de lípidos en la cabeza, el cuello y el iris. Como su nombre lo indica, se presentan en la infancia y por lo general en las niñas quienes regularmente manifiestan una ausencia de eliminación renal causando sepsis urinaria e inflamaciones en los riñones dando lugar a orinas turbias y fétidas. Sin embargo, estas características son extremadamente raras y por lo tanto no se hace necesario el efectuar exámenes periódicos de sangre u óseos.
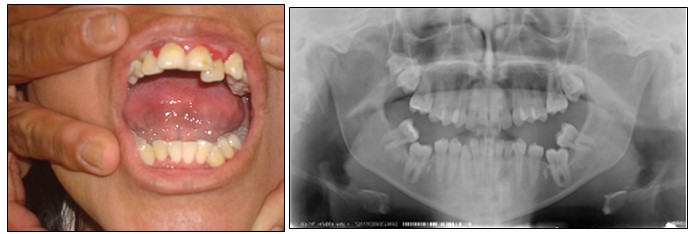
Tan solo a un 4 a 7% de los pacientes afectados con NF1 se les ha detectado manifestaciones orales. Todo tipo de tejidos orales, consistentes o suaves, han sido afectados por tumores siendo la lengua el lugar más común.
Los hallazgos más comunes son: neurofibromas orales, papilas fungiformes alargadas, lesiones intra óseas, canales alveolares inferiores anchos, foramen mandibular alargado; apiñamiento dental, tal como presenta la paciente de la imagen 30.
Los niños con NF-1 pueden padecer de migraña caracterizada por una cefalea continua o punzante, nauseas y dolores abdominales. Dichos infantes deben ser sometidos a cuidadosos exámenes físicos y neurológicos para excluir cualquier otra causa.
Los síntomas de problemas neurológicos, craneales o intracraneales pueden ser fácilmente detectados. Cambios en la vista, en la audición, el gusto o el habla; mareos súbitos sin causa aparente, desequilibrio, debilidad muscular, cambios de sensibilidad en cualquier área del cuerpo; cambios de comportamiento mental inexplicados, epilepsia, imposibilidad de mover ambos ojos completamente, dolores de cabeza fuertes, palpitaciones, sudoración, dolor de pecho y en el abdomen, nerviosismo, irritabilidad, incremento en el apetito, pérdida de peso, ataques.
Las lesiones endócrinas en los sistemas vasculares y gastrointestinales figuran también entre las características de la NF-1. En tales casos suelen presentarse constipaciones, cólicos abdominales y hemorragias gastrointestinales en el lugar donde se localiza uno o varios neurofibromas intestinales difusos. Estos son causantes de anemias graves y de enfermedades intestinales. Por lo general no se desarrollan en la infancia.
Finalmente, algunos pacientes han considerado los cambios climáticos, especialmente de calor a frío, como agravantes de algunas manifestaciones tales como el dolor o la rigidez. Otros sienten la adherencia de una banda pegada al cuerpo estirándose y produciendo aguijonazos. Un grupo de científicos estudia la teoría sobre si existe en el organismo una pérdida en los niveles de potasio, esta conlleva a un exceso de calcio el cual posiblemente activa señales intracelulares afectando células vecinas. Tales ideas no han sido clínicamente comprobadas.
Tumores cerebrales asociados a NF1.– Si bien los niños con NF1 están en riesgo de desarrollar un tumor cerebral de bajo grado que involucra el nervio óptico llamado glioma de la vía óptica (OPG), no se sabe cuál es la mejor manera de predecir qué niño desarrollará una OPG o quién requerirá tratamiento. Además, no está claro si algunos grupos de niños con gliomas ópticos asociados a NF1 tienen un mayor riesgo de pérdida de la visión o fracaso del tratamiento.
Como parte de un consorcio internacional, el director del Centro NF, David H. Gutmann, MD, PhD, y sus colegas están estudiando el impacto de la quimioterapia en la visión en niños con NF1. Estas investigaciones tienen como objetivo identificar los factores de riesgo para la pérdida de visión en niños con OPG. Además, el Dr. Gutmann y sus colegas han encabezado un estudio de múltiples investigadores para definir las alteraciones genéticas y genómicas en los tumores cerebrales de bajo grado (gliomas) pediátricos NF1 relevantes para mejorar el manejo del paciente. Finalmente, los investigadores del Centro NF de la Universidad de Washington han desarrollado criterios uniformes para clasificar los tumores cerebrales en niños con NF1. Estos estudios han llevado a una mayor apreciación del espectro de tumores cerebrales en NF1 importante para optimizar el cuidado de los individuos afectados
Cáncer (malignización).- Esta es probablemente la complicación que genera más preocupación en las personas involucradas con la NF-1, pacientes y médicos. En realidad, el riesgo de desarrollar cáncer que está definitivamente relacionado con la NF-1 es bajo (alrededor del 5%). Los dos tipos de cáncer particularmente asociados con la NF son los «tumores embrionarios» (tumores en la infancia que aparecen en tejidos embrionarios primitivos) y neurosarcomas (tumores malignos de los nervios, generados por cambios en el comportamiento de un neurofibroma preexistente).
Los tumores malignos responden mejor al tratamiento si se los encuentra cuando son muy pequeños. Cuando un paciente con NF-1 observa que un bulto le crece rápidamente (incrementando su tamaño cada semana) o volviéndose muy doloroso, debe consultar a su médico y explicarle estos nuevos síntomas.
Diagnóstico.- Para diagnosticar un paciente afectado con NF-1 desde el punto de vista clínico debe cumplir unos criterios diagnósticos y está estipulado que deben cumplir DOS o más de los que se detallan a continuación:
- – Seis o más manchas color «café con leche» mayores de 5 mm de diámetro en pacientes antes de la pubertad y más de 15 mm si se miden después de la pubertad.
- – Dos ó más neurofibromas de cualquier tipo (subcutáneo, cutáneo o plexiforme).
- – Pecas o efélides en las axilas y/o en ingles.
- – Gliomas en vías ópticas.
- – Dos ó más nódulos de Lisch (hamartomas benignos del iris).
- – Una lesión ósea característica (como la escoliosis).
- – Pariente de primer grado afectado de NF 1 (padre o hermano)
Es importante mencionar que a pesar de no ser considerados como elementos diagnósticos, la NF-1 puede presentarse además de manera ocasional:
- – Macrocefalia,
- – Pubertad precoz,
- – Hipertensión arterial,
- – Alteraciones de la estatura,
- – Dificultades en el lenguaje,
- – Deformidades óseas, y otras.
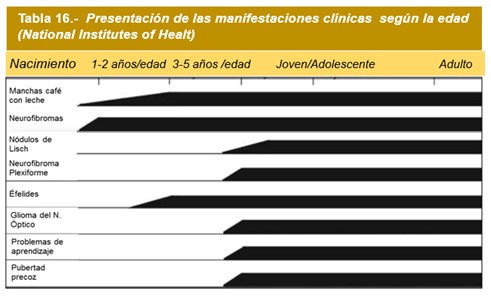
Por lo general, el orden de aparición es el siguiente: Manchas café con leche, pecas en las axilas, nódulos Lisch y neurofibromas. La Tabla 9 resume los criterios diagnósticos. Considerar además los hallazgos radiológicos, ecocardiográficos de neuroimágenes y genéticos.
Diagnóstico diferencial.– Síndrome de Leopard (lentigines, anomalías cardiacas, talla baja, ceguera), Melanosis cutánea (nevo piloso pigmentado gigante), Síndrome de Proteus. (sobrecrecimiento óseo, hamartomas subcutáneos, hierplasia giriforme de manos y pies), Síndrome de Klippel-Trénaunay-Weber (hemangioma con hemihipertrofia), Síndrome de Carney (nevo, mixoma auricular, neurofibromas mixoides y efélides), Lipomatosis múltiple (lipomas benignos múltiples), Síndrome de Bannayan-Riley-Ruvalcaba (lipomas múltiples y hemangiomas), Síndrome de McCune-Albright (displasia fibrosa poliostótica, disfunción endocrina y áreas de pigmentación), Síndrome de Sotos (gigantismo cerebral y pobre coordinación). Brevemente describiré algunos:
– Síndrome de Proteo (Véase en el Hipertexto).- Entidad caracterizada por hemihipertrofia, tumores subcutáneos y macrodactília. Descrita en 1979 por Cohen y Hayden. Debido a que el fenotipo de estos pacientes es cambiante con la edad, Wiedeman le dio el nombre de Proteus en honor al Dios griego que cambiaba su forma para evitar su captura. Este síndrome es un complejo trastorno hamartomatoso de causa desconocida, probablemente relacionado con la mutación de un gen en el mosaicismo somático.
Las alteraciones asociadas con el síndrome de Proteus pueden ser: anormalidades en el crecimiento (sobrecrecimiento asimétrico y desproporcionado del cuerpo, macrodactilia, aumento de la talla, macrocefalia), alteraciones cutáneas y subcutáneas (engrosamiento de la piel, nevus epidérmico, lipomatosis, linfagiomas y hemangiomas), afecciones óseas (hemihipertrofia, defectos de angulación de las rodillas, escoliosis, luxación de la cadera) y oculares (exostosis periorbital, tumor epibulbar, cataratas, miopía, nistagmus, ptosis, estrabismo y microftalmía). Otras anomalías que pueden encontrarse, ocasionalmente, son: embolia pulmonar, enfermedad pulmonar quística, lesiones urogenitales, cardiomiopatía hipertrófica, malformaciones cerebrales y vasculares.
En la mayoría de los casos las características hamartomatosas del síndrome se hacen evidentes sobre el año de vida, aunque pueden estar presentes en el nacimiento. Son progresivas durante toda la infancia y suelen cesar después de la pubertad. Esta entidad es polimórfica en cuanto a la localización de sus manifestaciones y su simetría y también lo es en cuanto al momento de su aparición; presenta pleomorfismo cronológico. Existe el riesgo de muerte prematura en los individuos afectados debido a la trombosis venosa profunda y embolismo pulmonar causado por las malformaciones arterio-venosas que se asocian a este trastorno. A causa de exceso de peso y agrandamiento de las extremidades, la artritis y dolores musculares son muy comunes.
– Enfermedad de McCune-Albright (Véase en el Hipertexto),- Denominada también Displasia Fibrosa Poliostótica. La enfermedad de McCune-Albright (véase más adelante) tiene características similares a la NF1: Manifestaciones óseas y pigmentaciones en la piel, tales como: MCL nacimiento, desarrollos fibrosos anormales en los huesos los cuales pueden conllevar a deformaciones óseas y fracturas y en algunos casos, sordera, ceguera y pubertad precoz en mujeres. Las MCL en esta entidad suelen ser más grandes, de contornos más irregulares y tienden a respetar la línea media. Las MCL también se han relacionado con estenosis de la válvula pulmonar (síndrome de Watson), esclerosis tuberosa, síndrome LEOPARD y neoplasia endócrina múltiple (MEN), aunque también pueden encontrarse algunas de estas lesiones en personas normales.
-Déficit de vitamina D.- Algunas de las enfermedades del sistema óseo se deben a una deficiencia nutricional en vitamina D.
-Displasia o Neoplasia Endocrina Múltiple tipo 2B (MEN2B).- Neurinomas múltiples de mucosas, anomalías musculo esqueléticas, feocromocitomas, diversas manifestaciones endocrinológicas.
-Neurofibromatosis tipo 2.- Neuromas acústicos, sordera, catarata posterior capsular, retinopatía pigmentaria, gliomas retinales, schwannomas.
Tratamientos.- Dianas terapéuticas emergentes para la NF Tipo 1.
Foto histórica: Grupo de Investigación para la Neurofibromatosis Tipo 1. Alumnos de la Cátedra de Medicina-Interna de la Universidad de Guayaquil-Ecuador, año 2014.

Bibliografía
- Recklinghausen Von FD. Uber die Multiplen Fibrome der Haut und Ihre Beziehung zu den Multiplen Neuromen. Berlin: A. Hirschwald, 1882.
- Virchow R. Ueber die Reform der patologischen und therapeutischen Anschauungen durch die mikroskopischen Untersuchungen. Virchow Arch Pathol Anat Physiol Klin Med. 1847; 1:345-9.
- Huson SM, Compston DA, Clark P, Harper PS. A genetic study of Von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989; 26:704-11
- Valero MC, Pascual-Castroviejo I, Velasco E, Moreno F, Hernandez-Chico C. Identification of de novo deletions at the NF1 gene: no preferential paternal origin and phenotypic analysis of patients. Hum Genet 1977; 99: 720-6.
- Barker D, Wright E, Nguyen K, Canon L, Fain P, Goldgar D, et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science. 1987; 236: 1100-2.
- Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Faryniarz AG, Chao MV, et al. Genetic linkage of von Recklinghausen neurofibromatosis to the nerve growth factor gene. Cell. 1987; 49:589-94.
- Malhotra R, Ratner N. Localization of neurofibromin to keratinocytes and melanocytes in developing rat and human skin. J Invest Dermatol. 1994; 102:812-8. 8. Dasgupta B, Gutmann DH. Closing the GAP between mice a men. Curr Opin Genet Dev. 2003; 13:20-7.
- Dasgupta B, Gutmann DH. Closing the GAP between mice an men. Curr Opin Genet Dev. 2003; 13:20-7.
- Costa RM, Silva AJ. Mouse models of neurofibromatosis type 1: Bridging the GAP. Trends Mol Med. 2003; 9:19-23
- Virginia C. Williams, John Lucas, Michael A. Babcock, David H. Gutmann, Bruce Korf , Bernard L. Maria. Neurofibromatosis Tipo 1 Revisitado. Pediatrics Volume 123, Number 1, January 2009.