Nota: Las imágenes cuentan con el consentimiento informado del familiar.
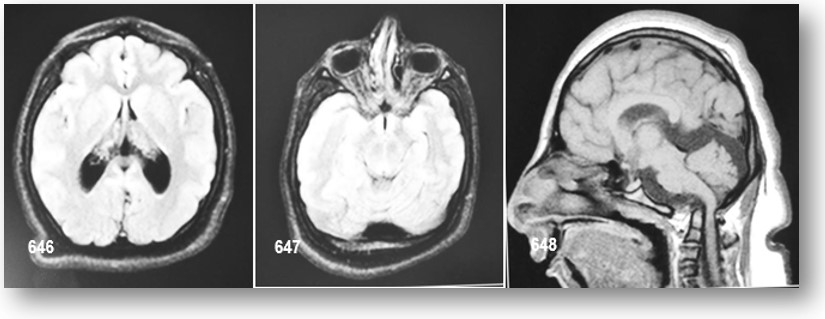
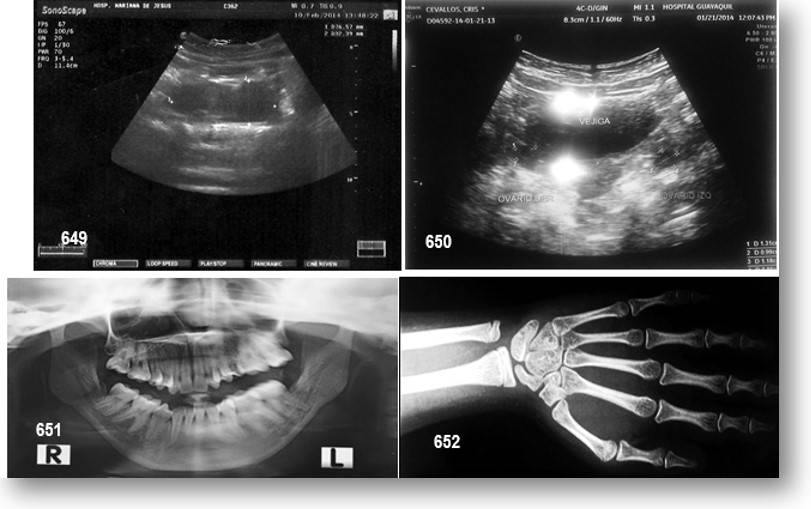
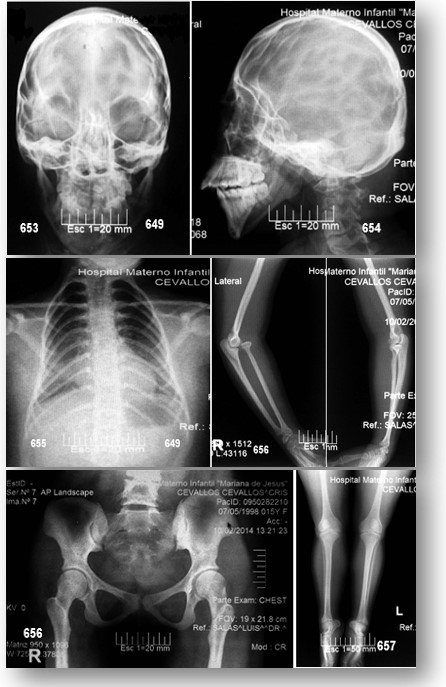
La RMN del cerebro efectuada actualmente no reporta cambios importantes (véase imágenes y reportes), ni otra realizada hace 2 años atrás. La radiografía de tórax no muestra cambios importantes, excepto la presencia de 11 costillas; un estudio ecográfico pélvico revelo riñón derecho ectópico (pélvico), con hipoplasia severa del izquierdo (esbozo); véase imágenes y reportes, además evidencia la presencia de útero en anteversión y ovarios de tamaño acorde a su enanismo proporcional. Serie ósea, sin interés patológico digno de mención. Edad ósea.
El estudio hormonal reveló valores elevados de ACTH, testosterona, dehidrotestosterona y 17 Hidroxiprogesterona (véase examen de laboratorio). Presentó un cariotipo femenino normal (46XX) y genitales internos femeninos normales en la ecografía pélvica.
Estudios paraclínicos (anormalidades en negrillas): Hemograma: Hematíes: 5.160.00 mm3; Hb: 15G/dl; Htco: 44.6%; Leucocitos: 8.350 mm3; Diferencial: normal; VCM: 86 fL; HCM: 29pg;CHCM: 34%; RDW:14%;MPV: 275.000mm3; Colesterol total: 188mg/dl; cHDL: 34,84 mg/dl; cLDL: 121mg/dl; LDL/HDL: 3.47; Índice de Castelli: 5.40 (riesgo elevado); Triglicéridos: 184 mg/dl; Proteínas Totales: 8.92 g/dl; S-Albumina: 5.58g/dl; S-Globulinas: 3.34g/dl; Índice A/G: 1.67 g/dl; Urea: 15 mg/dl; Creatinina: 0.53mg/dl; Acido úrico: 5.22 mg/dl; Bilirrubina total: 0,51mg/dl; Apo A1: 122mg/dl; Apo B: 136.30 mg/dl (60-133); Lp(a): 128,90 mg/dl (0.0-30.0); GOT: 35 UI/L; GPT: 49 UI/L; GammaGT: 120UI/L; Homocisteina: 6.87 umol/L; IgG: 166 mg/dl; IgA: 252 mg/dl; IgM: 127 mg/dl; Anticuerpos antireceptor TSH: 0,90 U/L; TSH: 2.17 uUI/ml; T4 L: 0,70 ng/dl; T4 Total: 6.10 ug/dl; T3 Total: 2.48 nmol/L; Dihidrotestosterona: 413 pg/dl (Referencias: mujeres: premeno:24-368; postmeno:10-181); Testosterona: 1.03 ng/ml; Cortisol 8am: 14,29 ug/dl; FSH: 5.21 mUI/ml (Referencias: FF:3.5-12.5; FPO: 5-16; FO: 4.7-21.5; FL: 1.7-7.7); LH: 10.95 mUI/ml (Referencias: FF: 2.4-12.6; FO: 14-95.6; FL: 1-58.5); Prolactina: 8.84 ng/ml; Progesterona: 0,33 ng/ml (Referencias: FF:0.3-1.1; FL: 1.8-2.11); 17-betaestradiol: 60 pg/ml (FF: 12.5-166;FL: 43.8-211;FO: 85.8-498); Estradiol libre en suero: <0.07 ng/ml; PTH: 40pg/mL; 17-Hidroxiprogesterona: 3.03 ng/ml (Referencia: niñas 1-13 años: 0.07-1.53); ACTH: 93pg/ml; GH: 0,91 ng/ml; IGF1: 487 ng/ml (Referencia: 432 la media); IGFBP3: 8.38 ug/ml; Glucosa: 100mg/dl; Insulina plasmática: 73.65; HOMA-IR: 18,17 uUI/L (Referencia: normal: 2.10-2.70; RI: 4.30-5.20; DM con RI: 8.30-9.50). Elemental de orina: normal.
Estudio del corazón. Ecocardiograma unibidimensional: No se realizó.
Estudio citogenético (Figura 40): Cariotipo en sangre periférica: Se ha efectuado el cultivo de sangre periférica y realizado el recuento de 20 metafases que presentan 46 cromosomas. Con la técnica de bandas G, ni los cromosomas del tipo XX, ni los autosomas presentan una morfología sensiblemente diferente al cariotipo femenino normal. Resultado: Cariotipo 46 XX.
 Estudios genéticos: Secuenciación genética: No se realizó.
Estudios genéticos: Secuenciación genética: No se realizó.
INFORMACION BÁSICA.- Enanismo Primordial Tipo MOPD II (OMIM:210720/).- En la especie humana existen unos doscientos cincuenta tipos diferentes de síndromes que provocan enanismo (más de 200 sin nombre).
Enanismo Primordial es una rara forma de enanismo que se traduce en un menor tamaño corporal en todas las etapas de la vida a partir de antes de nacer. Más concretamente, el enanismo primordial es una categoría de diagnóstico, incluidos ciertos tipos de enanismo, en el cual los individuos son muy pequeños para su edad, incluso en un feto. La mayoría de las personas con enanismo primordial no son diagnosticadas hasta casi los 3 años de edad.
Se trata de una mutación identificada en el cromosoma 21, más en concreto en la localización 21q22.3, que afecta al gen PCNT, que se expresa a través de la proteína pericentrina, uno de los componentes principales del centrosoma (centro regulador de la progresión del ciclo celular). Esta mutación genética entraña una pérdida de función bialélica que da lugar a una incorrecta separación de los cromosomas durante la división celular.
La mayoría de los casos de baja estatura son causados por malformaciones esqueléticas o trastornos endocrinos. Los cinco subtipos de enanismo primordial son las formas más graves de la mayoría de los 200 tipos de enanismo, y se estima que sólo hay 100 personas en el mundo con el trastorno. Otras fuentes de la lista del número de personas actualmente afectadas viven en América del Norte (EUA).
Es raro que las personas afectadas por enanismo primordial vivan más allá de la edad de 30 años. En el caso de microcefalia en el enanismo primordial osteodisplasico (MOPD) tipo 2 puede incrementarse el riesgo de problemas vasculares, que pueden causar la muerte prematura.
El correcto diagnóstico de la EP no puede hacerse hasta que el niño tiene 5 años y se hace evidente que el niño tiene enanismo severo.
TIPOS DE ENANISMOS PRIMORDIAL. ENANISMO PRIMORDIAL OSTEODISPLÁSICO (MOPD) TIPO 1 y 3.- El tipo 1 tipos 1 (también llamado síndrome de Taybi-Linder) y el tipo 3 de enanismo microcefálico osteodisplásico primordial (MOPD; por su siglas en inglés) se caracterizan por: retraso de crecimiento intrauterino y postnatal, microcefalia, dismorfia facial, displasia esquelética, peso bajo al nacer y anomalías cerebrales. En un inicio, los tipos 1 y 3 de MOPD fueron descritos como dos entidades separadas, en base a criterios radiológicos (pequeñas diferencias en la estructura ósea de la pelvis y de los huesos largos). Sin embargo, las últimas investigaciones confirmaron que las dos formas representaban modos de expresión diferentes de un mismo síndrome. La prevalencia es desconocida, pero se han descrito en la literatura menos de 30 casos hasta el momento. La dismorfia facial se caracteriza por: nariz prominente con puente nasal plano, ojos saltones, frente huidiza y micrognatia. Otros signos característicos de la enfermedad son: cabello y cejas escasas, piel seca, miembros cortos y luxación de cadera y codos. Las manifestaciones neurológicas más frecuentes son: convulsiones y déficit intelectual; mientras que las anomalías cerebrales más frecuentes son: lisencefalia, hipoplasia de lóbulos frontales y agenesia del cuerpo calloso o del vermis cerebeloso. Los tipos 1 y 3 de MOPD se transmiten según el modo autosómico recesivo. A pesar de que el gen responsable de la enfermedad no se conoce, el mapeo de homocigotos ha permitido la identificación de una región génica candidata en el cromosoma 2q (2q14.2-q14.3). Los estudios histológicos sugieren que los dos tipos de MOPD están provocados por un defecto de la proliferación celular y de la diferenciación tisular. El diagnóstico se basa en el fenotipo clínico y radiológico, incluyendo como signos radiológicos característicos: acortamiento de los huesos tubulares, arqueamiento de los huesos largos y ampliación del espacio intervertebral. El diagnóstico diferencial incluye el MOPD de tipo 2 (ver este término) y otros síndrome asociados con enanismo primordial (como el síndrome de Seckel; ver este término). Se ha descrito el diagnóstico prenatal por ecografía alrededor de las 20 semanas de gestación en familias afectadas. El tratamiento es sólo sintomático. El pronóstico es grave, con muerte durante el primer año de vida constatada en la mayoría de los casos descritos.
Nos referiremos exclusivamente al tipo 2 de este grupo de nanismos osteodisplásicos primarios, debido a su parecido con las manifestaciones fenotípicas del síndrome de Seckel.
ENANISMO OSTEODISPLÁSICO PRIMORDIAL DWARFISMO DE TIPO 2 (MOPD 2).- Las características notables de MOPD 2 son: retardo severo del crecimiento intrauterino, retraso severo del crecimiento postnatal; tamaño de la cabeza relativamente proporcional al nacer que progresa a microcefalia verdadera y desproporcionada; desproporción progresiva de talla baja secundaria a acortamiento de los segmentos distales y medios de las extremidades; una displasia ósea
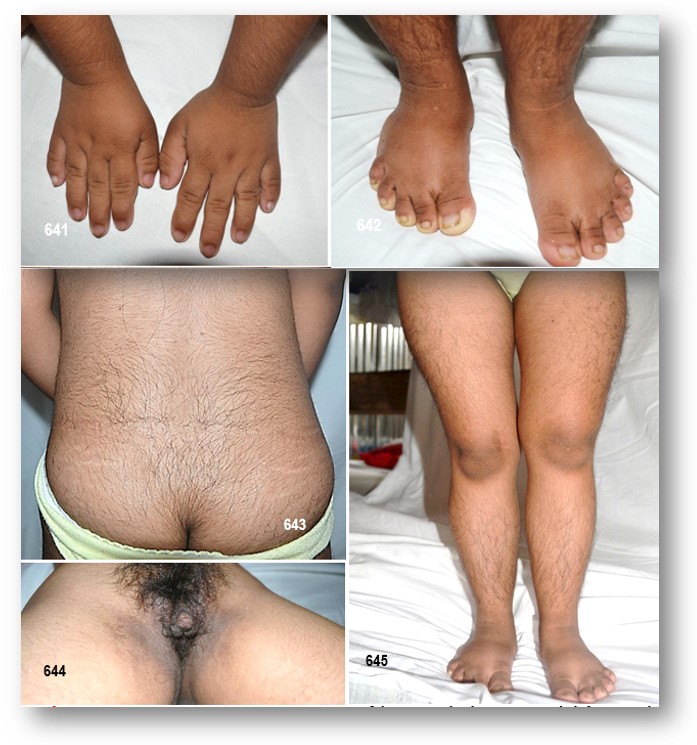
progresiva con cambios metafisiarios en las extremidades; retardo en el desarrollo de las epífisis; progresivas deformidades por luxaciones o subluxaciones de las rodillas, cabezas radiales y caderas (displasia de la cadera); otras anomalías esqueléticas en el MOPD 2 incluyen: el desarrollo anormal de las articulaciones, adelgazamiento de los huesos de los brazos y las piernas, escoliosis, y los huesos de la muñeca acortados. Rasgos faciales inusuales, incluyendo una nariz prominente (corno), con la punta hipoplástica, ala nasal delgada y la columela que queda debajo del ala nasal. Mejillas prominentes. Ojos que parecen grandes en la lactancia y la primera infancia, las orejas, que son proporcionales, ligeramente displásicas y generalmente ausencia del lóbulo; voz alta y chillona (nasal) como consecuencia de la estenosis subglótica; boca anormalmente pequeña, y a menudo dentición displásica (microdontia) o faltan (adontia); son gentiles y de carácter agradable, extrovertidos, y de personalidad sociables.
La visión de futuro suele estar comprometida (hipermetropía). Con el tiempo, los individuos afectados pueden desarrollar áreas de coloración de la piel anormalmente clara u oscura (pigmentación). Obesidad troncal a menudo desarrollan con el tiempo. Hiperinsulinismo por severa resistencia a la misma. Ovarios poliquísticos también ha sido reportado. Algunas personas parecen ser más susceptibles a las infecciones.
Todos los rasgos faciales que hemos descrito suelen cambiar con el tiempo.
En las personas con el crecimiento de la cabeza MOPD 2 ralentiza en el tiempo, los individuos afectados tienen un tamaño del cerebro adulto comparable a la de un bebé de 3 meses de edad. Sin embargo, el desarrollo intelectual es típicamente normal.
La RMN de la región hipotálamo-hipofisiaria es sugestiva de hipoplasia hipofisiaria. Un número de personas afectadas han desarrollado la dilatación de las arterias del SNC descritos diversamente como aneurismas y la enfermedad de Moya Moya. La patogenia típica moyamoya comienza con estrechamiento de los vasos en la base del cerebro, en la arteria carótida interna supraclinoideo, segmentos de la arteria cerebral anterior o cerebral media. El estrechamiento puede predominar inicialmente en un lado, progresa a estenosis bilateral, con la consiguiente oclusión de los vasos y la formación de colaterales. Estas anomalías vasculares son a menudo tratables, aunque aumentan el riesgo de derrame cerebral y reducen la esperanza de vida de los individuos afectados. Estos cambios vasculares pueden ser potencialmente mortales, incluso en los primeros años a causa de la ruptura, hemorragia del SNC, y los accidentes cerebrovasculares. Existe variabilidad entre los individuos afectados, incluso dentro de la misma familia.
Los problemas de crecimiento en MOPD 2 son primordiales, es decir, que comienzan antes del nacimiento, con las personas afectadas que muestran el crecimiento prenatal lento (retraso del crecimiento intrauterino). Después del nacimiento, los individuos afectados continúan creciendo a un ritmo muy lento. La altura final del adulto de las personas con esta condición va desde 20 pulgadas a 40 pulgadas.
MOPDII parece ser una enfermedad poco frecuente, aunque su prevalencia es desconocida.
Las causas del MOPD 2 son mutaciones en el gen de la pericentrina (PCNT), que codifica la síntesis de una proteína llamada igual.
La PCNT es un gen que proporciona instrucciones a la pericentrina dentro de las células. La PCNT se encuentra en los centrosomas, que son parte de los cromosomas, y juegan un papel en la división celular y el ensamblaje de los microtúbulos. Los microtúbulos son fibras que ayudan a las células mantener su forma, ayudar en el proceso de la división celular, y son esenciales para el transporte de materiales dentro de las células. La PCNT actúa como una proteína de anclaje, asegurando otras proteínas en el centrosoma. A través de sus interacciones con estas proteínas, pericentrina juega un papel en la regulación del ciclo celular, que es la forma de la célula de replicarse a sí mismo en un paso a paso de manera organizada.
Las mutaciones de la PCNT de genes conducen a la producción de una proteína no funcional pericentrina que no pueden anclar otras proteínas en el centrosoma. Como resultado, los centrosomas no pueden ensamblar correctamente los microtúbulos, lo que conduce a la interrupción del ciclo celular y la división celular. La división celular alterado provoca una reducción en la producción de células, mientras que la interrupción del ciclo celular puede conducir a la muerte celular. Esta reducción global en el número de células conduce a huesos cortos, microcefalia, y los otros signos y síntomas de MOPD 2.
Esta condición se hereda en un patrón autosómico recesivo, lo que significa que ambas copias del gen en cada célula tienen mutaciones. Los padres de un individuo con un trastorno autosómico recesivo llevan cada uno una copia del gen mutado, pero por lo general no muestran signos y síntomas de la enfermedad.
Pacientes afectos con la MOPD 2 fueron diagnosticados de síndrome de Seckel, pero durante la última década se han identificado 10 genes presentes en diferentes síndromes de enanismo primordial.
Diagnóstico diferencial.- El diagnóstico diferencial de los enanismos osteodisplásicos primordial se lo hacen con los otros dwarfismos:
– Síndrome de Seckel (OMIM 210600): En las personas con Síndrome de Seckel se observa que la microcefalia en muchos de ellos también sufren de escoliosis, luxación de cadera, retraso en la edad ósea, luxación de la cabeza radial, y convulsiones.
– Enanismo Osteodisplásico Primordial, de tipo 1 (MOPD 1) (210710): Esta forma de enanismo primordial es a menudo abreviado como MOPD II. En el interior de su cerebro, el cuerpo calloso es a menudo poco desarrollado (agenesia del cuerpo calloso) y se sabe que tienen convulsiones y apnea. La delgadez de pelo también es común, incluyendo el cuero cabelludo, el cabello, las pestañas y las cejas. El esqueleto es de vértebras cortas, alargadas clavículas, se inclinación de fémures y el desplazamiento de la cadera. Al igual que las personas con Síndrome de Seckel también a menudo tienen microcefalia.
– Enanismo Osteodisplásico Primordial, tipo 2 (MOPD 2) ( 210720): Los que tienen a menudo tienen más MOPD 2 problemas médicos que los otros tipos, como una voz chillona, microdoncia, muy separados entre sí los dientes primarios, los patrones de sueño pobres (en los primeros años), retraso mental, enfermedad frecuente, problemas respiratorios, problemas de alimentación, hiperactividad, problemas de la visión, los aneurismas cerebrales, y no responden a la terapia hormonal, ya que el enanismo primordial no es causado por una falta de hormona del crecimiento. Después de revisar los rayos X también se encuentra que muchos tienen articulaciones dislocadas, la escoliosis y la edad ósea retrasada, así como microcefalia. No van a llegar al tamaño de un recién nacido medio hasta que estén entre las edades de 3-5.
Los efectos morfológicos de esta mutación corresponden a un tipo de enanismo de carácter hereditario, conocido como MOPD 2, que origina individuos adultos de una talla no superior a un metro, cuyo cerebro presenta un tamaño similar al de un bebé de tres meses -su circunferencia no supera los 40 centímetros- pero con una capacidad intelectual y cognitiva muy cercana a la inteligencia de una persona normal.
- Mutación en el gen de pericentrina, igual defecto en el síndrome de Seckel.
- Mutación del enanismo osteodisplásico tipo 1 y 3, RNA, U4atac small nuclear (U12-dependent splicing) – RNU4ATAC
– Síndrome de Russel-Silver (180860: La altura final de las personas con Síndrome de Russel-Silver a menudo excede la altura de otras personas con enanismo primordial, y todos en conjunto son muy diferentes. Algunas propiedades de las personas que tienen Síndrome de Russel-Silver son palmeados dedos de los pies, no descienden los testículos (en varones), el tono muscular débil, retraso en la edad ósea, labio superior delgado, hipospadias, la voz de tono alto, mentón pequeño, retraso en el cierre de la fontanela, hipoglucemia, y una frente ancha. Sus cabezas pueden parecer de forma triangular y grande para su tamaño pequeño cuerpo.
– Síndrome de Gorlin-Meier (224690): es conocido por sus víctimas con orejas pequeñas y rótulas no. También se le encuentran curvas en las clavículas, costillas muy delgadas, y la dislocación del codo. Al igual que el síndrome de Russel-Silver, por lo general superior a la altura de las personas con Síndrome de Seckel y MODP 2.
No se ha registrado ningún tratamiento eficaz para el enanismo primordial. La falta de crecimiento normal en el trastorno no se debe a una deficiencia de la hormona del crecimiento, como en el enanismo hipofisario. La administración de hormona del crecimiento, por lo tanto, tiene poco o ningún efecto sobre el crecimiento de la persona con enanismo primordial. En enero de 2008, se publico que las mutaciones en el gen pericentrina (PCNT) se encontró que causa enanismo primordial.
Discusión.- Los Enanismos Microcefálicos Osteodisplásicos Primordial (EMODP) es un desorden autosómico recesivo (similar al SCKL), que posee características distintivas, aunque a veces muy sutiles que trae conflictos nosológicos. Su diagnóstico como el de todos los EMODP, es clínico, sin embargo suelen superponerse entre ellos.
El estudio de la secuenciación génica es importante para filiar con más seguridad el genotipo.
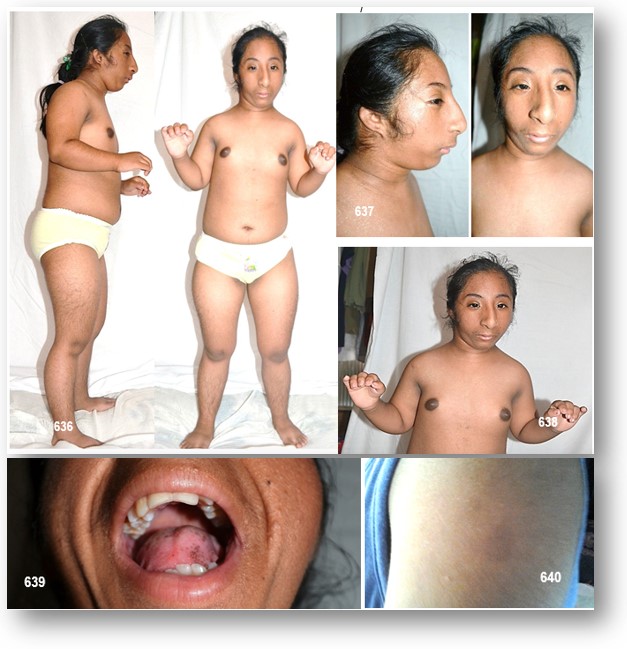
En caso que presentamos motivo de esta monografía, las características clínicas-semiológicas observadas, se fueron definiendo de manera progresiva, es así que nos encontramos con una paciente de características displásicas muy peculiares, el enanismo como denominador común, y con anomalías craneofaciales a simple como la microcefalia, nariz prominente, ojos pequeños asociado a otras malformaciones y compromiso de la agudeza visual. Las orejas de implantación baja con ausencia de lóbulos, como se ha descrito en los EMODP, con micro y retrognatia. La asimetría facial destacaba su cara. El enanismo como era de esperarse es desproporcionado a expensas de los miembros superiores. Obesidad troncal aparente con miembros inferiores seudomusculosos. Nos sorprendió que frente a un cráneo tan pequeño su capacidad intelectual se veía menoscaba por una lenta respuesta a las preguntas más sencillas como responder al preguntarle su nombre, y que al escuchar su voz chillona nos invitó a pensar aún más que estábamos frente a un EMODP de tipo 2, y no a un Síndrome de Seckel. El resto de las malformaciones corporales presentes simplemente forman parte del contexto de esta entidad. Para mayor información véase la tabla 50.
La presencia de un hiperclitoris (elemento este constante en la EMOP, incluyendo el SCKL), conjuntamente con otros signos de virilización, nos sorprendieron cuando al revisar los exámenes de laboratorio en especial el perfil hormonal nos invitó a pensar que estábamos frente a una hiperplasia suprarrenal congénita subyacente.
El desarrollo sexual de caracteres sexuales secundarios estaba de acorde a la maduración de la Escala de Tanner. Sin embargo, cabe la posibilidad de que la abuela de la paciente confundiera fechas. Lo que si era claro es que estábamos frente a una amenorrea primaria con genitales normales y un perfil de gonadotropinas similares, aunque dentro del contexto de una hiperplasia suprarrenal congénita. Sin embargo, si nos llamó la atención de sobremanera al examen ginecológico vello púbico abundante, espeso, grueso y rizados, en la vulva la presencia de placas hipertróficas, hiperpigmentadas e hiperqueratósicas que confunde estructuras labiales, estas placas de aspecto sucio, muy sugestivas de acantosis nigricans no eran otra cosa que labios menores hipertrofiados, los mismos que no pueden diferenciarse; no se determina introito vaginal, pero si hipertrofia del clítoris.
Estos hallazgos sugestivos de virilización, son como consecuencia de una hiperplasia suprarrenal congénita, los cambios pigmentarios a acantosis nigricans y las alteraciones a nivel de labios menores a acrocordones de una dermodisplasia verrucciforme, como consecuencia de la insulinorresistencia severa (véase el HOMA-IR). El hirsutismo, así como las manchas hipercrómicas (café con leche), son también muy sugestivos de un EMODP de tipo 2.
Conclusiones.- En todos los casos de EMODP se identifican mutaciones de la PCNT. Es más, el análisis retrospectivo de pacientes diagnosticados como portadores del SCKL con mutación también en el gen de la PCNT, sugiere que todos ellos pertenecen al espectro de los EMODP. Sin embargo, pacientes con EMODP 2 pueden ser diagnosticados erróneamente como portadores del SCKL, basados en la ausencia de manifestaciones severas del esqueleto y en su examen final la estatura >110 centímetro que normalmente excluye el diagnóstico de EMODP 2.
El espectro clínico del EMODP 2 es heterogéneo (al igual que el SCKL) y sufre de la definición inconstante y el conflicto nosológico en la literatura médica y por consiguiente en la práctica, como lo ha constatado nuestro grupo de estudio, al describir el primer caso documentado del SCKL en Ecuador. De hecho, el SCKL se ha usado a menudo como un término genérico usado para el enanismo primordial, sin que concurra un diagnóstico más específico.
Recientemente se ha informado dos casos del SCKL con anomalías intracraneales sugestivas de EMODP 2. Semejantemente, pacientes con displasias óseas muy sugestivos de ser portadores del SCKL se ha informado de que en realidad padecen de EMODP 2.
Nosotros sugerimos que el espectro clínico del EMODP 2 es más amplio que el previamente definido. Sin embargo, en todos los pacientes con mutaciones de la PCNT, investigadores de crédito resumen que de forma consistente se observa en el EMODP 2: elementos clínicos constantes como los rasgos faciales distintos; retraso en el crecimiento <5 DS y microcefalia <5 DS; apacibles de carácter, y algo muy esencial, el retraso mental ausente.

Las manifestaciones esqueléticas incluyen displasia de la cadera que va del cuello femoral corto a la coxa vara severa, condensación carpiana, y adelgazamiento metafisiario de los huesos largos. Otros rasgos sugestivos del EMODP 2 y no del SCKL, incluyen: anomalías vasculares y cutis marmorata; la voz alta, aguda y chillona por estenosis subglótica (?); la microdontia; el hiperinsulinismo (acantosis nigricans), y las anomalías de pigmentación de la piel con áreas de hipo e hiperpigmentación, estos últimos encontrados en nuestra paciente.
Por último, y al tenor del espectro clínico de nuestro caso, dejamos en claro que las manifestaciones de virilización descritas en el contexto de los EMODP, incluyendo el SCKL, serian manifestaciones de una hiperplasia suprarrenal congénita subyacente, ya que no existe otra justificación etiológica para su presencia.
El objetivo de esta publicación es de dar a conocer la posibilidad de que los pacientes con EMODP que presentan simultáneamente estos dos síndromes, sugiriere un mecanismo etiopatogénico común, por lo menos en las manifestaciones de virilización.
Foto histórica. – Grupo para la investigación del Enanismo Microcefálico Osteodisplásico Primordial de Tipo II. Universidad de Guayaquil-Ecuador. Con mis alumnos del grupo 7 de la Cátedra de Medicina-Interna, año 2017.
Nota: El Editor principal del HIPERTEXTO, Dr. Francisco A. Tama Viteri sosteniendo en sus brazos a la paciente portadora del Síndrome de Seckel, y a su lado de pies, la paciente portadora del MOPD Tipo II.

Bibliografía
- Faivre L, Cormier-Daire V. Seckel syndrome. Orphanet encyclopedia 2005
- Seckel, H. P. G. Bird-headed Dwarfs: Studies in Developmental Anthropology Including Human Proportions. Springfield, Ill.: Charles C Thomas (pub.) 1960.
- Science; DOI: 10.1126/ science.1151174)
- Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II): Natural History and Clínicasl Findings. Judith G. Hall, Christina Flora, Charles I. Scott Jr, Richard M. Pauli, and Kimi I. Tanaka. American Journal of Medical Genetics 2004;130A:55–72.
- Bobabilla-Morales L., Corona-Rivera A., Corona-Rivera J.R., Buenrostro C., García-Cobián T.A., Corona-Rivera E., Cantú-Garza J.M. and García-Cruz D. 2003. Chromosome Instability Induce In Vitro UIT Mitomycin C in Five Seckel Syndrome Patients. Am. J. Hum. Genet.123A:148-152
- Featherstone LS, Sherman SJ, Quigg MH. Prenatal diagnosis of Seckel syndrome.J Ultrasound Med1996;15: 85-8.
- Tama Viteri FA; Sanchez Carvajal MD. Síndrome de Seckel. Enanismo “Cabeza de Pájaro”. Presentación del Primer Caso Descrito en Ecuador. Revista de Medicina de la Facultad de CC.MM de la Universidad de Guayaquil 2013; 16:3, Supl 1.
- Tama Viteri FA; Sanchez Carvajal MD. Enanismo Micriocefalico Osteodisplasico Primordial Tipo II. Asociado a Hiperplasia Suprarrenal Congenita Virilizante.Presentacion del Primer Caso Descrito en Ecuador. Revista de Medicina de la Facultad de CC.MM de la Universidad de Guayaquil 2014; 17:1, Supl 2.