INFORMACIÓN BÁSICA: Síndrome de Waardenburg (OMIM: 193500/ORPHA:896/CIE-10:): Un síndrome genético que se presenta desde el desarrollo del embrión humano, presentando alteraciones en las células de pigmento, con consecuencias de despigmentación en parches en piel, ojos y pelo, así como sordera en un porcentaje de los casos. Su reconocimiento será importante para su pronóstico. Se trata de un síndrome genético debido a defectos en el desarrollo del tejido del sistema nervioso en el embrión humano, que se presenta con defectos en la pigmentación, pudiendo afectar a la piel, el pelo y/o la coloración del iris principalmente. Es la causa más común de la sordera sensorial, de severidad variante y de tipo hereditario.
Clasificación o variantes Clínicas del Síndrome de Waardenburg:
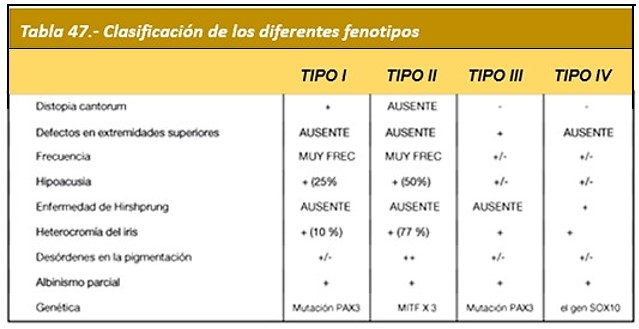
Tipo I: Afecta a los melanocitos. Presenta despigmentación en parches. Relacionado al gen PAX3. No presenta tan frecuentemente sordera.
Anomalías faciales:
- – Puente nasal ancho.
- – Ausencia del ángulo fronto- nasal.
- – Hipoplasia de las alas de la nariz.
- – Labio superior en «arco de cupido».
- – Labio inferior grueso.
- – Mandíbula prominente.
- – Mechón de cabello blanco (40%).
- – Conjunción de las cejas (sinofridia).
- – Labio leporino.
- – Distopía cantorum: disposición anómala de los ojos: desplazamiento lateral de los ángulos internos de los ojos.
Anomalías visuales:
- – Heterocromía del iris (ojos de diferente color).
- – Microftalmia (pequeñez anormal de los ojos) y cataratas; no es muy frecuente.
- -Anomalías auditivas:
- – Sordera en el 20-25% de los pacientes.
- – No hay distopía cantorum.
Tipo II: Se hereda de forma Autosómica Dominante. Además de las características del Tipo I, se presenta frecuencia elevada de sordera. Relacionado al gen WS2A del cromosoma 3. Se ha asociado además al gen mutado MITF el cual está involucrado en una falla en los melanocitos. Esta mutación a veces no se encuentra en familias.
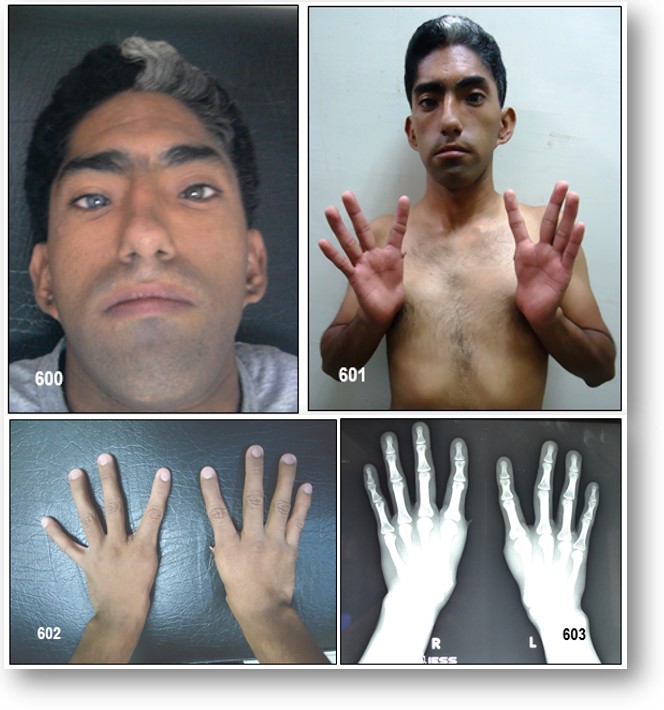
Tipo III (Síndrome de Klein-Waardenburg) (OMIM:148820/ORPHA:896/CIE-10:E70.3): Es conocido como Síndrome de Klein-Waardenburg. Relacionado a una variante del gen PAX3. Además de los hallazgos faciales y auditivos descritos en el tipo I, hay afectación bilateral de las extremidades superiores: deformidades por contracturas en flexión, fusión de los huesos del carpo y sindactilia (adherencia de dos o más dedos entre sí). Se pueden encontrar también disminución del desarrollo muscular, contracturas articulares, malformaciones esqueléticas.
Se considera el tipo más grave. El compromiso esquelético de las manos de mi paciente me llevo a esta variante.
Tipo IV: (Síndrome de Shah-Waardenburg): Se presentan las características del tipo II, pero incluye además la Enfermedad de Hirschprung. La enfermedad de Hirschprung es un trastorno hereditario con movimientos musculares poco funcionales en el intestino, ocasionando obstrucción del intestino desde el nacimiento. Mutación genes: EDN3; EDNRB; o SOX 10.
Las alteraciones pigmentarias del iris varían y pueden incluir:
- a) Heterocromía del iris completa (dos ojos de color diferente)
- b) Heterocromía parcial o segmentaria (segmentos de pigmentación azul o marrón en uno o ambos ojos)
- c) Ojos azules hipoplásicos (característicamente azul brillante en ambos ojos)
La pérdida de audición en síndrome Waarderburg tipo 1:
Se observa en aproximadamente el 60% de los individuos, es congénito, neurosensorial, típicamente no progresivo, puede ser unilateral o bilateral.
Criterios de diagnóstico para Waardenburg tipo 1 y tipo 2, propuestos por el Consorcio de Waardenburg.
- – Tipo 1: dos criterios mayores o uno mayor más dos menores.
- – Tipo 2: dos criterios mayores (con un criterio mayor de un familiar de primer grado afectado y este debe mostrar dos criterios mayores).
Importante valoración por especialista en genética clínica si es posible.
Genética.- Mutaciones en varios genes con locus en diferentes cromosomas se han establecido como causantes de los 4 tipos de SW, que además se diferencian en sus características fenotípicas y patrones de herencia ( Tabla 1).
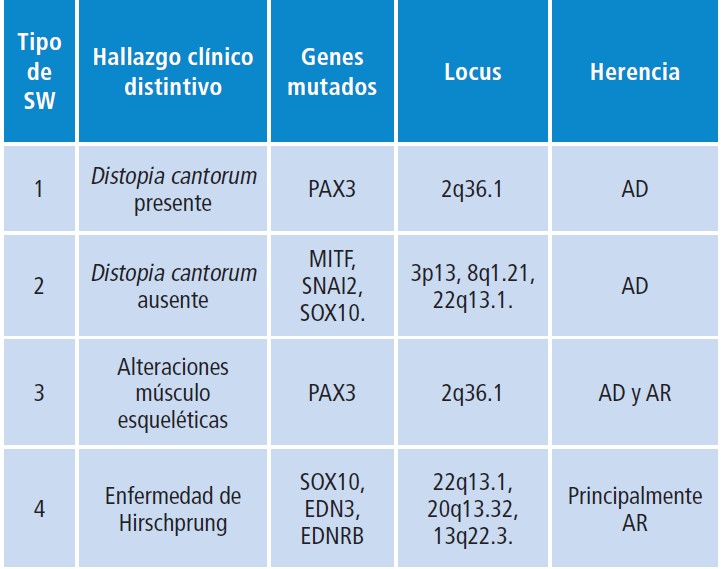
Se ha encontrado un vínculo entre la presentación del SW y la presencia de mutaciones en el en PAX 3, el cual se ubica en el cromosoma 2. La pérdida de la función de este gen se atribuye responsable de la creación de series de melanocitos defectuosos, células defectuosas del sistema nervioso en el oído interno y a errores en la información de células que lleva al exceso de tejido que ocasiona el exceso de separación entre los ojos y los defectos en miembros superiores. Las afectadas en el gen PAX3, que presentan herencia autosómica dominante, son responsables de los tipos 1 y 3 del SW. Este gen juega un papel importante en la embriogénesis, codifica un factor de transcripción que se une al ADN y es fundamental en el mantenimiento de las células madre pluripotenciales, las selecciones de linajes celulares, fallar, migración, apoptosis e inhibición de la diferenciación terminal, Notaron especialmente los derivados de la cresta neural, incluyendo los melanocitos en donde su expresión es determinante para una adecuada migración y diferenciación.
El impacto que tienen las mutaciones en el gen PAX3 sobre el fenotipo aún no se encuentra totalmente claro, dado que la expresión fenotípica es extremadamente variable, incluso dentro de una misma familia como la descrita en este informe. Aquí encontramos tres generaciones examinadas con un fenotipo diferente, dado que la madre de los gemelos tenia hipoacusia, pero no sus hijos ni su madre; para lo cual se ha propuesto que existe una posible interacción con genes modificados que explicaría que a pesar de ser la misma mutación no tendrán resultados clínicos equivalentes.
Se ha propuesto que el desarrollo de la región frontonasal es sensible a la actividad del factor de transcripción codificado por el gen PAX3, pues los defectos en esa región ocurren en todos los afectados con receptores en este gen, explicando así cómo la totalidad de casos de SW tipo 1 tienen distopia cantorum —definida cualitativamente como el desplazamiento lateral del canto ocular interno y cuantitativamente como un IW mayor de 1.95 —; de igual forma, los defectos pigmentarios y el grado de pérdida de la audición varía entre los individuos afectados, donde las alteraciones del iris, la poliosis y la hipoacusia pueden tener expresividad variable, mientras que la distopia cantorum suele estar presente en la totalidad de los casos denunciados.
Los genes de la familia PAX también participan en el desarrollo de las extremidades. El SW tipo 3, que también presenta distopia cantorum acompañado de alteraciones musculo-esqueléticas producto de cambios en el gen PAX3, se manifiesta cuando las alteraciones en dicho gen son heredadas en homocigosis.
El SW se puede diagnosticar y clasificar clínicamente utilizando los criterios establecidos por el Consorcio Internacional para el Estudio del Síndrome de Waardenburg. El SW tipo 1 se caracteriza, entre otras hallazgos clínicos, por presentar distopia cantorum , mientras que en el tipo 2 está ausente ( Tablas 1); el tipo 3, también llamado síndrome de Klein Waardenburg, además de la distopia cantorum , se asocia con alteraciones musculares-esqueléticas en miembros superiores y el tipo 4 o síndrome de Shah-Waardenburg presenta como característica distintiva una asociación con la enfermedad de Hirschsprung.
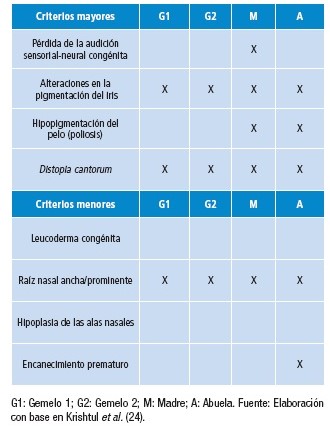
Dadas las diferencias fenotípicas y patrones de herencia específicos en los diferentes tipos de SW ( Tabla 1), se puede hacer un diagnóstico diferencial en ausencia de pruebas moleculares. El diagnóstico del SW tipo 1 se realiza cuando están presentes dos criterios mayores más un criterio menor o un criterio mayor más dos menores. El SW tipo 2 se diagnostica cuando están presentes dos criterios mayores y hay ausencia de distopia cantorum .
En el heredograma de la familias reportadas con el SW se puede encontrar un patrón de herencia autosómico dominante de las características clásicas de SW y se descartaron los tipos 3 y 4 teniendo en cuenta la ausencia de hipoplasia, sindactilia, contracturas u otras alteraciones musculo esqueléticas en miembros superiores y de signos y síntomas sugestivos de la enfermedad de Hirschprung.
En la búsqueda realizada en las bases de datos médicos, solo se encontraron dos publicaciones de gemelos monocigóticos con SW, ninguna de ellas después del año 2000. Esto demostró que si bien el SW es una enfermedad poco frecuente —1 por 40000 habitantes—, su diagnóstico en gemelos monocigóticos es aún más raro.
El oído se ve afectado en relación a los problemas pigmentarios, ya que los melanocitos son requeridos para los dos procesos. En el oído interno los melanocitos son necesarios en una capa llamada stria vascularis, para mantener una composición equilibrada de los iones y la endolinfa, una sustancia importante dentro del oído interno. En la ausencia de los melanocitos en esta zona, la consecuencia es la pérdida de la audición. Se han documentado diferentes tipos del síndrome de Waardenburg hasta el momento; los tipos I y II son los más comunes: El tipo de herencia es autosómico dominante. Este tipo de herencia es en la que el gen que está alterado es el dominante, invalidando otras copias del gen. De este modo, si uno de los genes de la enfermedad pasa al hijo, heredará la enfermedad. En familias con uno de los padres afectado, las posibilidades de heredar la enfermedad a los hijos son del 50% por cada embarazo. Los rasgos físicos suelen ser muy similares en una misma familia, aunque la gravedad de cada caso puede ser variable. Los pacientes con síndrome de Waardenburg presentan una gama variable de características físicas, dentro de las más frecuentes se encuentran:
- – Piebaldismo: mechón de pelo blanco, manchas despigmentadas en piel y despigmentación del iris.
- – Aumento de la separación entre los ojos.
- – Ojos pequeños, pueden ser de un azul muy pálido o ser uno sólo el que tenga color claro.
- – Párpados caídos
Se ha documentado una mayor incidencia de cataratas en algunos de estos pacientes. En una variante del síndrome pueden presentarse malformaciones de los brazos, presentando un desarrollo menor, además de rigidez en las articulaciones y malformaciones en los dedos. El síndrome de Waardenburg también se ha asociado con muchos trastornos de tipo congénito, como el intestino y la columna vertebral defectuosa, elevación de la escápula, alteraciones esqueléticas (ausencia de pulgares, por ej.) y el labio leporino.
Diagnóstico: se realiza en base a los siguientes criterios:
Criterios Mayores
- – Pérdida neurosensorial auditiva.
- – Anomalía en la pigmentación del iris (25% de los casos): heterocromia, iris bicolor, segmento de dos colores e iris azul zafiro brillante.
- – Alteración pigmentaria del pelo: mechón blanco en cráneo, barba y vello corporal (20-40% de los casos).
- – Labio leporino.
- – Distopia cantorum (aparente) es necesario medir el índice W (desplazamiento lateral del canto interno) un índice W superior de 1.95 es indicativo de distopia
– Familiares de primer grado afectados
Criterios Menores
- – Puente nasal ancho (75% de los casos).
- – Mayor separación entre los ojos (hipertelorismo) (10% de los casos).
- – Conjunción de las cejas (50% de los casos).
- – Canicie prematura, antes de los 30 años de edad (20-40% de los casos).
- – Decoloración de la piel (leucodermia).
Para hacer el diagnóstico deben hallarse 2 criterios mayores o 1 mayor y 2 menores.
Como métodos auxiliares de diagnóstico se plantean la audiometría, las pruebas genéticas, el tiempo tránsito intestinal y la biopsia de colon. El SW no existe tratamiento específico. Se debe prestar atención a cualquier deficiencia auditiva siendo posible que se necesiten audífonos, educación escolar apropiada y en algunos casos el implante coclear. A los pacientes con el tipo IV que sufren de estreñimiento se les indica una dieta particular y medicamentos que mantengan el movimiento intestinal. Una vez que corregidos los problemas de audición, la mayoría de las personas afectadas con SW pueden llevar una vida normal. Sin embargo, aquellas personas con las formas menos comunes de esta enfermedad pueden tener problemas adicionales, los cuales pueden afectar su pronóstico.
Las complicaciones incluyen pérdida auditiva, trastornos psicológicos o de autoestima relacionados con la apariencia, estreñimiento grave que requiere la extirpación de una parte del intestino grueso, aumento leve en el riesgo de rabdomiosarcoma, ligera disminución del funcionamiento intelectual (posible pero inusual). En las mujeres embarazadas con mayor riesgo de tener hijos con SW, debido a la asociación de defectos en el cierre del tubo neural está recomendada la suplementación con ácido fólico.
Bibliografía
- Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet 1951;3:195-253.
- Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol 2006;72:326.
- A, Aliagaoglu C. Waardenburg syndrome type 1. Dermatol Online J 2006;12:21.
- Genetics Home Reference. (2016). Waardenburg syndrome. Obtenido de Genetics Home Reference.
- Waardenburg Syndrome. Andrew P. Read. St. Mary’s Hospital, Mahcester, England. Genetic Hearing Loss. Patrick J.
- Willems. Capítulo 6.