INFORMACIÓN BÁSICA: Síndromes de Enhlers-Danlos (SED) (OMIM:-/ORPHAN:98249/CIE-10:Q79.6). Los SED son un grupo de enfermedades que afectan el tejido conectivo, que apoya la piel, huesos, tendones, ligamentos, vasos sanguíneos y otros órganos. Los signos y síntomas del síndrome varían según el tipo de SED y van desde juntas (articulaciones) que son un poco sueltas a problemas muy graves. La mayoría de los tipos tienen articulaciones que se pueden mover más de lo normal (hipermovilidad) principalmente en el tipo llamado SED tipo hipermóvil (SED tipo III). Muchas personas con SED también tienen una piel suave y aterciopelada que es muy elástica y frágil. Las personas afectadas presentan moretones con cualquier golpe y en algunos tipos también hay cicatrización anormal. Algunos tipos del síndrome, principalmente el tipo vascular y tipo cifoscoliosis, pueden tener problemas graves y que pueden ser mortales. El SED puede ser causado por mutaciones en diferentes genes. La forma de herencia varía según el tipo. No tiene cura por el momento. El tratamiento depende de los problemas que se presenten y puede incluir fisioterapia y drogas para aliviar los síntomas.
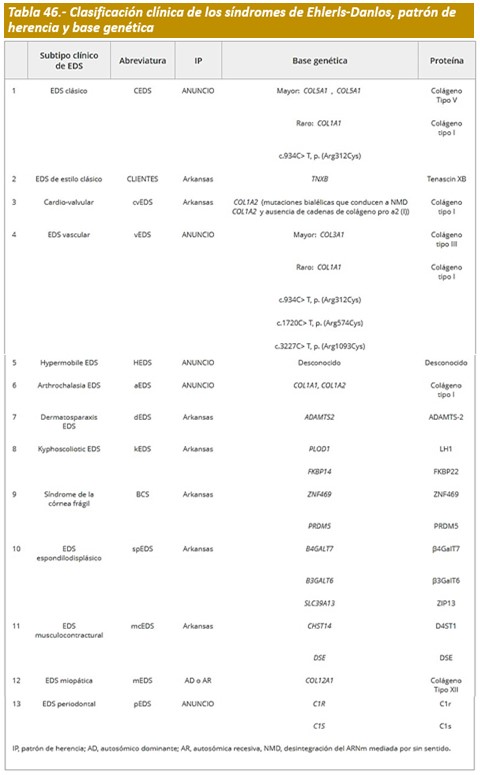
Clasificación.- Los SED son un grupo clínico y genéticamente heterogéneo de trastornos hereditarios del tejido conectivo (HCTD) caracterizados por hipermovilidad articular, hiperextensibilidad de la piel y fragilidad tisular. En las últimas dos décadas, la Nosología de Villefranche, que delineó seis subtipos, ha sido ampliamente utilizada como el estándar para el diagnóstico clínico de SED. Para la mayoría de estos subtipos, se han identificado mutaciones en genes que codifican colágeno o en genes que codifican enzimas modificadoras de colágeno. Desde su publicación en 1998, se ha descrito un amplio espectro de nuevos subtipos de SED, y se han identificado mutaciones en una serie de genes novedosos. El Consorcio Internacional EDS del 2017 propone una clasificación SED revisada, que reconoce 13 subtipos (Tabla 42). Para cada uno de los subtipos, proponemos un conjunto de criterios clínicos que son sugerentes para el diagnóstico. En vista de la gran heterogeneidad genética y la variabilidad fenotípica de los subtipos de SED, y la superposición clínica entre los subtipos de SED, pero también con otros HCTD, el diagnóstico definitivo de todos los subtipos de SED, excepto el tipo hipermóvil, depende de la confirmación molecular con identificación de (a) variante (s) genética (es) causativa (s).
También revisamos los criterios clínicos para EDS hipermóvil con el fin de permitir una mejor distinción de otros trastornos de la hipermovilidad articular. Para satisfacer las necesidades de investigación, también proponemos un esquema patogenético, que reagrupa subtipos SED para los cuales las proteínas causales funcionan dentro de la misma vía. Esperamos que la Clasificación Internacional revisada de SED sirva como un nuevo estándar para el diagnóstico de SED y proporcionará un marco para futuros propósitos de investigación.
Funciones de los genes: El gen COL5A1 o COL5A2 proporciona instrucciones para hacer un componente del colágeno tipo V. Los colágenos son una familia de proteínas que fortalecen y sostienen muchos tejidos del cuerpo, como la piel, los ligamentos, los huesos, los tendones y los músculos.
Un componente del colágeno tipo V llamado cadena pro-α1 (V) se produce a partir del gen COL5A1. Los colágenos comienzan como moléculas de procolágeno similares a cuerdas que están formadas por tres cadenas. Dos combinaciones de cadenas pueden producir colágeno de tipo V: tres cadenas pro-α1 (V) o dos cadenas pro-α1 (V) y una cadena pro-α2 (V) (que se produce a partir del gen COL5A2.
Las moléculas de procolágeno de triple cadena son procesadas por enzimas fuera de la célula para crear colágeno maduro. Las moléculas de colágeno luego se organizan en fibrillas largas y delgadas con otra forma de colágeno, tipo I. El colágeno tipo V regula el ancho (diámetro) de esas fibrillas. Los estudios sugieren que el colágeno tipo V también controla el ensamblaje de otros tipos de colágeno en fibrillas en varios tejidos.
El gen COL1A1 proporciona instrucciones para hacer parte de una molécula grande llamada colágeno tipo I. Los colágenos son una familia de proteínas que fortalecen y sostienen muchos tejidos del cuerpo, como el cartílago, el hueso, el tendón, la piel y la parte blanca del ojo (la esclerótica). El colágeno tipo I es la forma más abundante de colágeno en el cuerpo humano.
Un componente del colágeno tipo I llamado cadena pro-α1 (I) se produce a partir del gen COL1A1. Los colágenos comienzan como moléculas de procolágeno similares a cuerdas que están formadas por tres cadenas. El colágeno de tipo I está compuesto de dos cadenas pro-α1 (I) y una cadena pro-α2 (I) (que se produce a partir del gen COL1A2.
Las moléculas de procolágeno de triple cadena son procesadas por enzimas fuera de la célula para crear colágeno maduro. Las moléculas de colágeno luego se organizan en fibrillas largas y delgadas que forman interacciones estables (enlaces cruzados) entre sí en los espacios entre las células. Los enlaces cruzados dan como resultado la formación de fibras de colágeno tipo I muy fuertes.
El gen COL1A2 proporciona instrucciones para hacer parte de una molécula grande llamada colágeno tipo I. Los colágenos son una familia de proteínas que fortalecen y sostienen muchos tejidos del cuerpo, como el cartílago, el hueso, el tendón, la piel y la parte blanca del ojo (la esclerótica). El colágeno tipo I es la forma más abundante de colágeno en el cuerpo humano.
Un componente del colágeno tipo I llamado cadena pro-α2 (I) se produce a partir del gen COL1A2. Los colágenos comienzan como moléculas de procolágeno similares a cuerdas que están formadas por tres cadenas. El colágeno de tipo I está compuesto de dos cadenas pro-α1 (I) (que se producen a partir del gen COL1A1 y una cadena pro-α2 (I).
Las moléculas de procolágeno de triple cadena son procesadas por enzimas fuera de la célula para crear colágeno maduro. Las moléculas de colágeno luego se organizan en fibrillas largas y delgadas que forman interacciones estables (enlaces cruzados) entre sí en los espacios entre las células. Los enlaces cruzados dan como resultado la formación de fibras de colágeno tipo I muy fuertes.
El gen ADAMTS2 proporciona instrucciones para producir una enzima que procesa varios tipos de moléculas de procolágeno. Los procolágenos son los precursores de los colágenos, que son moléculas complejas que se encuentran en los espacios entre las células que agregan fuerza, soporte y elasticidad a muchos tejidos del cuerpo. La enzima ADAMTS2 corta una cadena corta de bloques de construcción de proteínas (aminoácidos) en un extremo de los procolágenos. Este paso de recorte es necesario para que las moléculas de colágeno resultantes se ensamblen en fibrillas fuertes y delgadas.
El gen PLOD1 o FKBP14 proporciona instrucciones para producir una enzima llamada lisil hidroxilasa 1. Esta enzima modifica un aminoácido llamado lisina, que es uno de los componentes básicos utilizados para fabricar proteínas. Específicamente, la lisil hidroxilasa 1 convierte la lisina en una molécula similar, hidroxilisina, a través de una reacción química llamada hidroxilación. La hidroxilisina se encuentra comúnmente en los colágenos, que son moléculas complejas que proporcionan fuerza y soporte a muchos tejidos corporales.
La hidroxilisina es esencial para que las moléculas de colágeno formen interacciones estables, llamadas enlaces cruzados, entre sí en los espacios entre las células.
Los enlaces cruzados dan como resultado la formación de fibras de colágeno muy fuertes.
El gen TNXB proporciona instrucciones para producir una proteína llamada tenascina-X. Esta proteína juega un papel importante en la organización y el mantenimiento de la estructura de los tejidos que sostienen los músculos, las articulaciones, los órganos y la piel del cuerpo (tejidos conectivos). En particular, los estudios sugieren que ayuda a regular la producción y el ensamblaje de ciertos tipos de colágeno. Los colágenos son una familia de proteínas que fortalecen y sostienen los tejidos conectivos en todo el cuerpo. Tenascin-X también participa en la regulación de la estructura y la estabilidad de las fibras elásticas, que proporcionan flexibilidad y elasticidad a los tejidos conectivos.
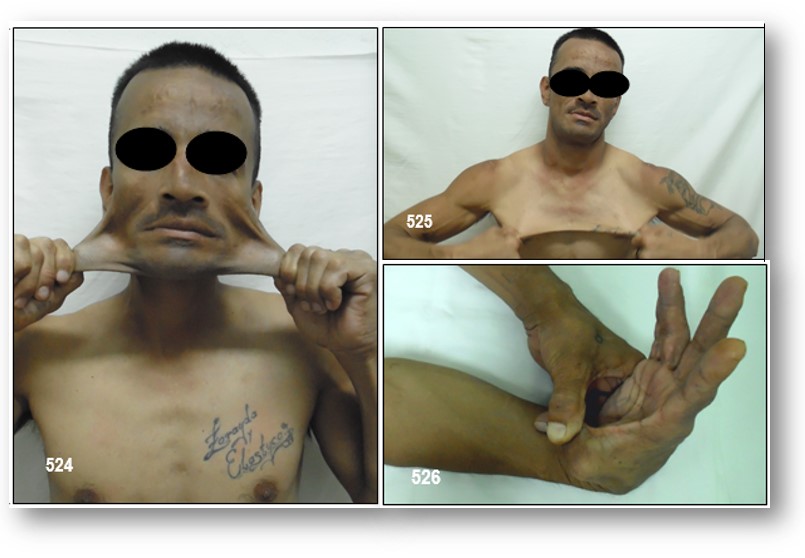
Síntomas.- Algunas personas pueden ser levemente afectados, mientras que otros pueden tener problemas graves y potencialmente mortales. Los principales síntomas están asociados con problemas en las articulaciones y la piel. Las señales y síntomas varían entre los diferentes tipos, pero de forma general pueden ser:
Hipermovilidad y/o hiperextensibilidad caracterizada por articulaciones que se mueven más allá del rango normal, que son sueltas o inestables y que se pueden dislocar más fácilmente:
- – Dolor en las articulaciones
- – Osteoartritis de inicio temprano
- – Piel suave, «aterciopelada»
- – Piel muy elástica
- – Piel frágil que se hiere fácilmente o moretones frecuentes
- – Mala cicatrización de heridas
- – Desarrollo de tumores “seudo moluscoides” (lesiones carnosas asociados con cicatrices en zonas de presión).
Otras señales y síntomas que pueden estar asociados con diferentes tipos de los síndromes de Ehlers-Danlos pueden ser:
- – Dolor crónico en los huesos y músculos que comienza en la juventud (principalmente en el tipo con hipermovilidad)
- – Fragilidad o ruptura de las arterias, de los intestinos o del útero (mas con el tipo vascular)
- – Curvatura anormal de la espalda (escoliosis) desde el nacimiento
- – Fragilidad de la esclerótica del ojo (mas con el tipo cifoscoliosis)
- – Tono muscular pobre (mas con el tipo Artrocalasia)
- – Prolapso de la válvula mitral
- – Enfermedad de las encías
Actualmente se reconocen 13 fenotipos diferentes:
– Síndrome de Ehlers-Danlos tipo clásico. El SED clásico se caracteriza por una marcada hiperelasticiad de las articulaciones, con gran susceptibilidad a esguinces, dislocaciones y subluxaciones. Puede incluso llegar a haber un desarrollo motor lento. Respecto a los órganos internos, podemos encontrar desde hernias de hiato a prolapsos rectales desde la niñez y se ve incrementado el riesgo de padecer hernias frecuentes. La anormalidad en las válvulas de corazón debido a que se encuentren deformadas llega a producir insuficiencia mitral. Es mucho más raro encontrar dilataciones de la aorta.
– Síndrome de Ehlers-Danlos por déficit de tenascina-X (tipo similar al –síndrome de Ehlers-Danlos tipo clásico). La Tenascin-X y especialmente su relación con el SED es en gran parte desconocido, por lo que en la literatura medica existente solo hay breves descripciones de este subtipo publicado hasta el momento y su superposición a otras enfermedades, en especial musculares.
– Síndrome de Ehlers-Danlos tipo cardiaco valvular. Se caracteriza porhipermovilidad, hiperextensibilidad de la piel y defectos de las válvulas del corazón.
– Síndrome de Ehlers-Danlos tipo hiperlaxo (hipermóvil). Es el segundo tipo más común. En esta variante las manifestaciones clásicas son la hipermovilidad de las articulaciones largas, como los codos y rodillas, y en muchos casos las de las manos y pies. Las dislocaciones son muy comunes, sobre todo las de los hombros y mandíbula. La hiperelasticidad de la piel hace que sea fácilmente dañable. El dolor crónico en casi todas las articulaciones hace que en los casos más severos, este síndrome puede llegar a ser inhabilitante.
– Síndrome de Ehlers-Danlos tipo vascular (SEDV). Poco frecuente, es el más grave de todos los subtipos ya que como característica principal engloba la ruptura de arteria u órganos. La piel llega a ser tan transparente que permite visualizar las venas. Existe tal grado de fragilidad capilar que puede llegar a romper los capilares al aplicar una mínima presión. La ruptura de vasos sanguíneos del intestino o del útero, cosa que aparece alrededor de los 30-40 años, es la principal causa de muerte en estos pacientes.
Existe un gran riesgo de sufrir hemorragias inesperadas durante el embarazo si se padece este subtipo. La hipermovilidad de las articulaciones, en este caso se restringe únicamente a los dedos de las manos. En este tipo de SED sí que encontramos un diagnóstico fiable, ya que gracias a una biopsia de la piel podemos identificar si hay una mutación en el gen COL3A1, lo que indica positivo para el SEDV.
– Síndrome de Ehlers-Danlos tipo cifoscoliosis. Este tipo de SED se caracteriza por presentar escoliosis congénita, que se incrementa conforme crece el paciente. Causa retraso en el desarrollo motor de los niños y en adultos puede provocar la incapacidad para caminar sobre los 20 a 30 años. También pueden presentase características del Síndrome de Marfan.
– Síndrome de Ehlers-Danlos tipo artrocalásico. La característica principal es la luxación congénita de la cadera. Esto va acompañado de un bajo tono muscular y ligera osteopenia.
– Síndrome de Ehlers-Danlos tipo dermatosparaxis. Este fenotipo tiene una severa fragilidad de la piel y por consiguiente constantemente se daña. Pueden aparecer hernias musculares o inguinales en algunos casos. Es el tipo menos común, y es el único tipo que se hereda de forma autosómica recesiva.
– Síndrome de la córnea frágil (antiguamente llamado tipo VIB). Este fenotipo se caracteriza por escleróticas azules, rotura corneal después de un traumatismo menor, queratocono o queratoglobus, hiperelasticidad de la piel e hipermovilidad articular.
– Síndrome de Ehlers-Danlos tipo displásico espondiloqueiral. Este fenotipo se caracteriza al nacer por una hipotonía muscular grave (que con frecuencia requiere un estudio neuromuscular invasivo), cifoscoliosis que es progresiva, hipermovilidad articular intensa y luxaciones, e hiperelasticidad marcada de la piel. Además, hay fragilidad de la piel con cicatrices anormales, osteopenia sin tendencia a las fracturas, a menudo un hábito Marfanoide y microcornea, y ocasionalmente ruptura de las arterias y el globo ocular.
– Síndrome de Ehlers-Danlos tipo musculocontractural. En este fenotipo la debilidad muscular es la caracteristica, y ocurre cuando el esfuerzo muscular no produce una contracción o movimiento muscular normal. La fuerza muscular esta reducida, acompañado de debilidad muscular o músculos débiles.
– Síndrome de Ehlers-Danlos tipo periodontitis (antiguamente llamado tipo VIII). Los signos y síntomas son similares a los del tipo clásico. Los síntomas adicionales incluyen enfermedad de las encías y tejidos que rodean a los dientes locual resulta en una pérdida prematura de la dentadura.
– Síndrome de Ehlers-Danlos tipo miopático: caracterizado por tono muscular disminuido y/o atrofia muscular, que mejora con la edad, contracciones de las articulaciones de la rodilla, cadera y codo y por hipermovilidad de las articulaciones distales (como las de las muñecas, las manos o de los tobillos y pies). Causada por mutaciones en el gen COL12A1.
Esta clasificación puede verse con detalles en la página en línea de la Asociación Nacional del Síndrome de Ehlers Danlos e Hiperlaxitud. https://ehlersdanlos.org.es/síndromes-de-ehlers-danlos
La incidencia en la población de los distintos fenotipos de SED es la siguiente:
- El tipo Clásico afectaría a 1/20.000 personas.
- El tipo Hiperlaxitud a 1/5.000-20.000 personas.
- El tipo Vascular a 1/50.000-250.000 personas.
- El tipo Cifoescoliosis 1/100.000 personas.
- El tipo Artrocalasia se ha descrito en 30 personas en el mundo.
- El tipo Dermatoparaxis se ha descrito en 8 personas en el mundo. Cabe decir que un 50% de los pacientes no se pueden categorizar de forma clara en un único síndrome, tienen una mezcla de síntomas de tipos diferentes.
Causas.- Los SED son causados por mutaciónes en varios genes diferentes dependiendo del tipo específico: ADAMTS2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, PLOD1 y TNXB. En algunos casos no se conoce el defecto genético. Las mutaciones en estos genes cambian la estructura, la producción o la transformación de colágeno o proteínas que se relacionan con el colágeno.
El colágeno da estructura y resistencia a los tejidos conectivos en todo el cuerpo. Un defecto en el colágeno puede debilitar el tejido conectivo en la piel, los huesos, vasos sanguíneos, y órganos, resultado en las características de la enfermedad.
Herencia.- La forma de herencia de los SED varía según el tipo:
Autosómica dominante: El tipo clásico, el tipo artrocalásico, el tipo hipermóvil, el tipo periodontitis, y algunos casos del tipo miopático generalmente se heredan de esta manera.
Autosómica recesiva: El tipo dermatosparaxis, el tipo cifoscoliosis, el tipo vascular, el tipo cardíaco-vascular, el tipo musculocontractural, el tipo espondilo-displástico, el tipo de la córnea frágil, el tipo clásico-like, y algunos casos del tipo miopático son heredados de esta forma.
De los tipos más raros la herencia es autosómica recesiva en casi todos menos en:
– Autosómica dominante: El SED tipo hipermóvil familiar (antiguamente llamado tipo XI)
– Herencia ligada al cromosoma X: Forma variante de Ehlers-Danlos asociada a heterotopía periventricular nodular y SED ligado al X (tipo V).
Diagnóstico.- Los SED se diagnostican con las señales y síntomas que hay y con la historia médica completa y un examen físico que busca las pistas diagnósticas importantes.
Para algunos tipos como el tipo vascular, el tipo artrocalasia, y el tipo dermatosparaxis, se pueden hacer pruebas genéticas o pruebas bioquímicas (realizadas en cultivos de fibroblastos de la piel, para ver los defectos de las tipos del colágeno y que miden la actividad de las enzimas que están disminuidas debido a la mutación del gen específico para cada enzima) para confirmar el diagnóstico. El tipo cifoscoliosis se puede identificar mediante un ensayo de enzima en la orina (para ver la actividad de la enzima lisil-hidroxilase que se encuentra disminuida en este tipo) y con pruebas genéticas. El tipo hipermóvil se diagnostica con las señales y síntomas porque no hay un examen de laboratorio que los pueda confirmar.
Todos los tipos de SED comparten las siguientes características clínicas en diversos grados:
Características de la piel:
- – Piel muy elástica (hiperextensible), frágil o piel floja
- – Mala curación de heridas
- – Tendencia a tener moretones fácilmente
- – Cicatrices atróficas que son anchas y delgadas, conocidas como «cicatrices de papel de cigarrillos»
Criterios para el diagnóstico del SED “clásico”
Criterios mayores:
- – Hiperextensibilidad de la piel y cicatrices atróficas
- – Hipermovilidad articular generalizada (GJH)
Criterios menores:
- – Facilidad para la formación de moretones
- – Piel suave y pastosa
- – Fragilidad de la piel (o fractura traumática)
- – Pseudotumores moluscoides
- – Esferoides subcutáneos
- – Hernia (o historia)
- – Pliegues de Epicanto
Complicaciones de la hipermovilidad de las articulaciones (por ejemplo, esguinces, luxación / subluxación, dolor, pie plano flexible)
Antecedentes familiares de un familiar de primer grado que cumpla con los criterios clínicos.
Criterios mínimos sugestivos para el SED clásico
– Criterio mayor: hiperextensibilidad cutánea y cicatrización atrófica
Más:
– Otro criterio mayor: GJH (Hipermovilidad articular generalizada)
– y / o: al menos tres criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
También es común encontrar arrugas epicánticas y pápulas piezogénicas.
Características en otros lugares del cuerpo:
- – Fragilidad de los vasos sanguíneos
- – Articulaciones muy móviles (hipermóviles)
- – Propensión a luxaciones (lesiones en las articulaciones (dislocaciones) en las que los huesos salen de su posición) o subluxaciones (dislocación parcial) espontáneas
Los tipos definidos de los SED tienen sus características propias, aunque hay muchas señales y síntomas que se sobreponen y por lo tanto, hacer un diagnóstico clínico claro para un tipo específico puede ser difícil en algunos casos.
Para los pacientes que cumplen el conjunto de requisitos clínicos mínimos para un subtipo específico de los EDS, pero que no tienen acceso a la confirmación con el examen genético, o en el que se identifica uno o más mutaciones en uno de los genes específicos de un subtipo, o en los que no se identifican mutaciones en ninguno de los genes específicos de un subtipo, puede realizarse un «diagnóstico clínico provisional» de un subtipo de los SED y los pacientes deben seguirse clínicamente.
Vea la lista de laboratorios que ofrecen el examen de genética para los EDS ofrecida por Genetic Testing Registry. La mayoría de las veces los laboratorios no aceptan contacto directo con los pacientes y sus familias solamente con un profesional de la salud.
El profesional de genética puede orientar para saber si se necesita hacer el examen genético.
Diagnóstico.- Las estrategias de diagnóstico molecular deberían basarse en las tecnologías NGS, que ofrecen la posibilidad de secuenciación paralela de múltiples genes. La resecuenciación dirigida de un panel de genes, por ejemplo, COL5A1, COL5A2, COL1A1 y COL1A2, es un enfoque de tiempo y costo efectivo para el diagnóstico molecular del EDS genéticamente heterogéneo. Cuando no se identifica mutación (o en caso de una enfermedad autosómica recesiva, solo una mutación), este enfoque se debe complementar con una estrategia de detección de variante numérica de copia (CNV) para identificar grandes deleciones o duplicaciones, por ejemplo, amplificación de sonda dependiente de ligadura múltiple (MLPA), qPCR o análisis de matriz dirigida. Alternativamente, o en una segunda fase, se puede usar la secuenciación del exoma completo (WES) o la secuenciación del genoma completo (WGS) y las técnicas de secuenciación del ARN, con análisis de datos que inicialmente se centra en los genes de interés para un subtipo de SED dado. En ausencia de la identificación de una mutación causal, este enfoque permite expandir el análisis a otros genes dentro del genoma. Biopsia de piel: En esta prueba, se toma una pequeña muestra dela piel para observarla en el microscopio de alta potencia en donde se revelen las características anormalidades de la estructura del colágeno que sepuede ver en ciertos tipos de SED. Análisis deorina: Un análisis de orina puede confirmar o excluir el tipo xifoescoliosis.
Verificación del diagnóstico clínico.- El cribado molecular mediante resecuenciación dirigida de un panel de gen que incluye al menos los genes COL5A1, COL5A2, COL1A1 y COL1A2, o mediante WES o WGS, está indicado. Cuando no se identifica una mutación, este enfoque se debe complementar con una estrategia de detección de CNV para identificar grandes eliminaciones o duplicaciones.
En caso de falta de disponibilidad de pruebas genéticas, los hallazgos de microscopía electrónica de transmisión (TEM) de flores de colágeno en la biopsia cutánea pueden respaldar el diagnóstico clínico, pero no pueden confirmarlo.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que los tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden pasar desapercibidos por las técnicas moleculares estándar de diagnóstico; sin embargo, se deben considerar diagnósticos alternativos en ausencia de (a) mutación (es) COL5A1, COL5A2, COL1A1 o COL1A2.
Más del 90% de los pacientes con CEDS albergan una mutación heterocigótica en uno de los genes que codifican colágeno de tipo V (COL5A1 y COL5A2).
Bibliografía
- Ehlers-Danlos syndrome. Genetics Home Reference. November, 2015; http://ghr.nlm.nih.gov/condition/ehlers-danlos-syndrome.
- Pauker SP & Stoler J. Clínicasl manifestations and diagnosis of Ehlers-Danlos syndromes. UpToDate. February 22, 2016; http://www.uptodate.com/contents/clínicasl-manifestations-and-diagnosis-of-ehlers-danlos-syndromes.
- Pauker SP & Stoler J. Clínicasl manifestations and diagnosis of Ehlers-Danlos syndromes. UpToDate. February, 2016; http://www.uptodate.com/contents/clínicasl-manifestations-and-diagnosis-of-ehlers-danlos-syndromes.
- What is EDS?. Ehlers-Danlos National Foundation. 2012; http://www.ednf.org. Accessed 3/13/2015.
- EDS types. The Ehlers-Danlos Society. 2017; https://ehlers-danlos.com/eds-types/#mEDS.
- Malfait F, Francomano C, Byers P et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet. March, 2017; 175(1):8-26.
- Defendi GL. Genetics of Ehlers-Danlos Syndrome. Medscape Reference. August, 2015.