INFORMACIÓN BÁSICA.- Síndrome de Cornelia de Lange (SdCL) (OMIM 122470).- El SdCL es un desorden multisistémico con una expresión variable marcada por un dimorfismo facial característico, déficit intelectual de grado variable, retraso en el crecimiento que empieza antes del nacimiento (2º trimestre), manos y pies anormales (oligodactilia, o a veces incluso una amputación más grave, y braquimetacarpia constante del primer metacarpo) y otras malformaciones (corazón, riñón, etc.).
Se conoce como SdCL a un trastorno congénito caracterizado por un conjunto de anomalías físicas, especialmente faciales, asociadas a retraso mental de grado variable, cuya base genética y molecular es desconocida.
Fue descrito en 1933 por la doctora Cornelia de Lange tras estudiar a dos niñas 6 y 17 meses con grave retraso mental y del crecimiento, que asociaban una de rasgos malformativos muy similares que las hacían muy parecidas.
Sospechó una causa común y publicó su observación. Años antes en 1916 el doctor Brachmann había publicado los resultados de una autopsia de un niño con defectos severos de los cúbitos, las manos con un solo dedo y características clínicas del SdCL.
Frecuencia: Mundialmente la incidencia es de 1 caso por 10,000-50,000 nacidos vivos. En España el estudio de malformaciones congénitas ECEMC ha obtenido una prevalencia neonatal mínima (se diagnostican preferentemente las formas completas o severas) de 0,971100.000 (Martinez Frías, 1998). El riesgo de tener un segundo hijo con este síndrome es muy bajo, del 2 al 5%, aunque algunos autores dan un riesgo de recurrencia aún más bajo. En nuestro pais la Fundación Espejo ha reportado algunos casos aunque creo que estas son subestimaciones.
En cuanto al reparto por sexos existe un ligero predominio de mujeres (1,311) (Jackson, 1993).
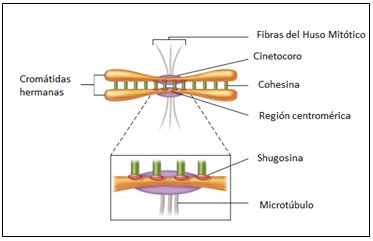
Causa.- La etiología de SdCL es desconocida. Aproximadamente el 99% de los casos son esporádicos, aunque ocasionalmente es transmitido con un patrón autosómico dominante. Se han reportado casos en gemelos con concordancia o discordancia, así como una posible herencia autosómica recesiva en algunas familias debido a mosaicismo o translocación 3q26:2q23. Las mutaciones asociadas con formas leves de la enfermedad en el gen SMC1L1 (también llamado SMC1A; Xp11.22-p11.21), asociado con una forma ligada al X de CdLS, y en el gen SMC3 (10q25). Recientemente se han identificado mutaciones heterocigóticas en un gen denominado NIPBL, el homólogo humano de la Drosophila melanogaster Nipped-B gen y aunque la función exacta de la delangina, proteína producto de NIPBL en humanos permanece desconocida, su homólogo en otras especies juegan un papel en el desarrollo, regulación y cohesión de cromátides hermanas.
El riesgo de recurrencia es de 0.5 a 1.5% si los padres no están afectados y del 50% si uno de los padres está afectado.
La mayoría de los casos son esporádicos, aunque se han visto casos en hermanos, en gemelos y en descendencia de padres consanguíneos, lo que puede sugerir una herencia autosómica recesiva.
También se han descrito casos en padre o madre e hijo la lo que sugiere una herencia autosómica dominante.
Por otra parte se ha visto un fenotipo similar en niños con duplicación del brazo largo del cromosoma 3 y en otras anomalías cromosómicas, así como tras la exposición a determinados teratógenos durante el embarazo (alcohol, valproico, dilantina).
También se ha sospechado que el origen pudiera estar en un segmento inestable de DNA que se amplifica en cada generación, para explicar el agravamiento de las manifestaciones clínicas en la segunda generación de una familia afecta.
Además muchos rasgos clínicos asemejan a la trisomía parcial 3q, lo que ha llevado a intentar identificar en esta región cromosómica la causa del SdCL, desde un punto de vista microcitogenético y molecular, dado que en la mayoría de los casos el cariotipo es normal.
La edad media de los padres no va a favor de neomutaciones dominantes, ya que la media no difiere de la existente en la población general. Con todo esto lo más probable es que la etiología sea heterogénea.
No se ha descrito diferencia de razas así como tampoco predilección en varones o hembras.
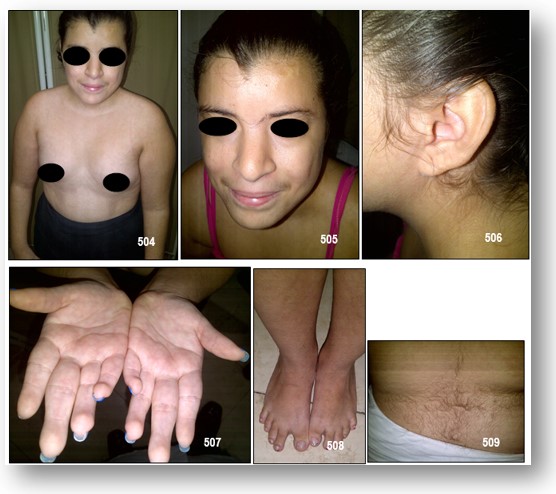
Características clínicas. Sin duda alguna son los más distintivos y en los que clínicamente se fundamenta el diagnóstico. La secuencia fotográfica adjunta resaltan los signos físicos más demostrativos destacando: talla baja, microcefalia, sinofris (cejas confluentes), pestañas largas, línea de pelo anterior y posterior de implantación baja, filtrum largo, labios delgados, narinas antevertidas, implantación baja de pabellones auriculares, micrognatia, cuello corto e hirsutismo. En nuestro caso tambien estuvieron presentes las malformaciones en extremidades superiores e inferiores, como la implatacion radial del pulgar, en las manos, y malformacviones de los dedos de los pies. En el 93% de los pacientes presentan micromelia. A nivel genital las mujeres afectadas presentan hipoplasia de labios mayores (57%) y los varones criptorquidia (73%) e hipospadias (33%). En el 40% de los casos se presentan anomalías del tracto urinario destacando: riñón en herradura, riñones hipoplásicos, ectopia renal, dilatación pielocalicial, quistes renales y reflujo vesicoureteral. Las Tablas 39 y 40 resumen los hallazgos clínicos.
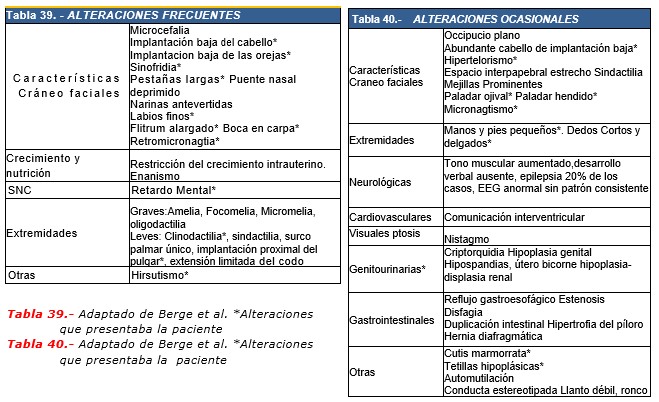
Se han descrito diversas cardiopatías congénitas sin predominio específico de alguna así como anomalías en huesos largos, incluyendo aplasia o hipoplasia de cabeza radial, fusión a nivel del codo y oligodactilia, sindactilia, acromilia, ectrodactilia y hasta focomelia. El 100% presenta edad ósea retardada y el 90% sordera neurosensorial.
En conclusión, el cuadro clínico es variable en intensidad, desde formas mayores o severas, a menores o poco sintomáticas.
Las mayores o tipo I se caracterizan por:
Hipocrecimiento global (98% de los afectados), con microcefalia, que se manifiesta ya al nacimiento (retraso de crecimiento intrauterino) y persiste en la vida adulta en los niños que sobreviven. Además se asocia a prematuridad hasta en un 3 1% de los casos.
Malformaciones externas acras: muy característicos defectos por reducción de extremidades que aparecen en un cuarta parte del total de los diagnosticados, manos y pies pequeños con cortedad de los dedos, en otros casos no tienen dedos o están malformados.
Rasgos faciales primitivos:
- – Sinofridia o sinofrisis (cejijuntos) presente en el 99 % de los individuos lo cual hace que sea el rasgo más distintivo.
- – Nariz pequeña y respingona con los orificios antevertidos, 88 % de los afectados.
- – Hirsutismo que puede ser generalizado 78 %.
- – Pestañas largas 99 %.
- – Labios finos 94 %.
- – Filtro prominente 94 %.
- – Angiomas.
Otras malformaciones:
- – Genitales: criptorquidia en el 73 % de los varones e hipoplasia genital en el 57 %.
- – Cardiacas.
- – Digestivas.
- – Reno-urológicas: reflujo vesico-ureteral en el 12%.
- – Oculares: defectos importantes en la visión en al menos un 50 %, miopía, ptosis, nistagmus.
- – Auditivas.
Limitaciones en la movilidad articular sobre todo codos en el 64% e implantación proximal del pulgar en el 72% de los casos, clinodactilia del 5 dedo. Deficiencia mental que se manifiesta al inicio como retraso en el desarrollo psicomotor. En general el grado se relaciona de forma directa con la intensidad del resto de los datos clínicos. El cociente intelectual varía entre 30 y 86, con una media de 53.
Anomalías en los dermatoglifos o pliegues y líneas de las manos:
- – Surco palmar de cuatro dedos 5 1 %.
- – Surco único de flexión en el 5″ dedo.
- – Patrón tenar: figuras en esa región.
- – Aumento de incidencia de bucles radiales digitales en dedos distintos del 2″.
- – Hipoplasia hipotenar con imagen de displasia arrosariada como signo de inmadurez.
- – Trirradio axial en posición media o distal.
- – Bucle de apertura dista1 o posición oblicua en 4″ espacio interdigital (Barr 197 1, Preus 1972, Schaumann B y Alter M 1976, Opitz 1985).
Las formas menores o tipo II son las que no muestran defectos de las extremidades y su inteligencia y fenotipo general están poco afectados. Habitualmente se diagnostican fuera del periodo neonatal.
El tipo III o fenocopia muestra una gran variabilidad clínica, los rasgos faciales son similares pero la expresión es parcial y a diferencia de los tipos I y II, con frecuencia se asocia a anomalías cromosómicas o a exposición a teratógenos.
Características psicomotoras.- Los que padecen este síndrome en ocasiones presentan asociado retraso psicomotor, observándose en la adquisición de las Iiabilidades de la coordinación, de la actividad mental y muscular. Puede observarse también retraso mental de nivel leve a severo.
En relación con los trastornos del comportamiento, ADAC (2003), señala que no se han realizado estudios abundantes sobre este tema, pero ha querido verse un carácter propio de las personas con SdCL que se caracterizaría por:
- – Poca habilidad para las relaciones sociales.
- – Comportamiento repetitivo y estereotipado.
- – Poca expresión facial de las emociones.
En pocas ocasiones se ha comprobado un comportamiento autoagresivo, pero incluso en los individuos con tipo 2 de SdCL, existe un perfil de personalidad rígido y muy de acuerdo con un medio ambiente muy estructurado.
En la investigación más importante en este síndrome, la revisión clínica de 310 individuos realizada por Jackson y colaboradores (1993) con el soporte de la Cornelia de Lange Syndrome Foundation, citada por ejemplo por el Club del filodendro (2003), aparecen datos relacionados con el desarrollo evolutivo.
Las puntuaciones obtenidas con algunos tests en niños con SdCL son estas:

En la siguiente tabla se compara la población en general con las edades en las que alcanzan algunos hitos del desarrollo, en el 50% de los casos, los niños con SdCL.

Los informes de la Cornelia de Lange Síndrome Foundation (CdLSF), Inc., matizan estos datos del siguiente modo:
El CI promedio de las personas con SdCL supone un retraso mental de leve a moderado para muchas personas con el síndrome, en lugar de los informes previos que iban de retraso moderado a severo.
En cuanto al desarrollo, el 25 % de los niños con SdCL presentan retraso en este aspecto. Los retrasos mayores se observan en el área de la comprensión oral y en el lenguaje expresivo. Pero los ámbitos en donde aparece un desarrollo más ajustado son los referidos a la memoria visual y espacial y a la organización perceptiva, con las posibilidades de aprendizaje que en estas áreas brinda actualmente la informática.
Lo que más se destaca es que todas estas personas aprenden a comunicarse y progresan a lo largo de su vida.
Lenguaje y habla.- Es uno de los aspectos indispensables. Los niños con SdCL pueden presentar retraso en el lenguaje o dificultad para entender los matices del lenguaje. En ocasiones es debido a que sufren apraxia oral-motriz. Es decir, tienen dificultades para producir los movimientos necesarios para hablar. Esto ocurre con más facilidad cuando se encuentran en ambientes escolares.
Características conductuales.- La persona típica SdCL puede ser descrita como hipersensible e irregular. La hipersensibilidad hace que el niño reaccione fuertemente al estímulo común y que la reacción continúe tiempo después que el estímulo se terminó. Por otro lado, en algunos niños sus rutinas de comportamiento en las áreas de alimentación, sueño y aspectos emocionales son irregulares.
Diagnóstico y evaluación.- En el momento actual no existe ninguna prueba definitiva para el diagnóstico; debe hacerse basándose en los datos clínicos.
Para personas expertas existen pocas dificultades para el diagnóstico de los casos clásicos. Los signos más valiosos son los faciales, sobre todo la sinofridia (98 %), pestañas largas (99 %), filtro largo, labios finos y comisuras bucales hacia abajo (94 %). Esto unido al retraso mental y pondoestatural, ofrece un diagnóstico con pocas dudas.
El problema del diagnóstico aparece fundamentalmente en los casos leves, es decir en los tipos II y III dónde tanto el retraso mental como el pondoestatural son menos frecuentes y marcados y los rasgos faciales están atenuados. A veces no aparecen malformaciones en las extremidades.
El diagnóstico prenatal es posible en algún hermano de un caso conocido, en el que existan anomalías de los miembros, microcefalia y retraso de crecimiento intrauterino visibles en las ecografías prenatales.
Pronóstico.- La mortalidad es alta, hasta tres veces la de la población general en algunas series (Beck, 1985). Las causas más frecuentes fueron respiratorias (neumonía, crisis de apnea, aspiración masiva de alimento al pulmón, displasia broncopulmonar …), seguidas por las malformaciones cardiacas y digestivas. En los que se realizó autopsia, el hecho más significativo en el cerebro fue la microcefalia. Durante la revisión de Jackson citada anteriormente tuvo lugar el fallecimiento de 14 niños con SdCL. Todos los casos fueron formas graves. 3 murieron por crisis de apnea, 3 por enfermedad cardiaca, 3 de aspiración masiva de alimento al pulmón, 2 por vólvulo intestinal, 2 en el postoperatorio por causa inexplicable, 1 por displasia broncopulmonar grave.
Tratamiento.- Durante el periodo de recién nacido y lactante presentan dificultad para la alimentación (77 %) que junto al reflujo gastroesofágico (48 %) agrava el problema del hipocrecimiento. El reflujo gastroesofágico requiere un tratamiento precoz y efectivo que en ocasiones debe de ser quirúrgico.
En un 25% de los casos existe patología respiratoria recidivante, en algunos casos directamente relacionados con el reflujo.
Es frecuente también la patología oftalmológica: miopía, ptosis, estrabismo, nistagmus. Su tratamiento está dificultado porque los niños en general no toleran las gafas.
En un 20% de los afectados se ha descrito además sordera de grado variable. En estos casos es preciso un diagnóstico precoz ya que puede condicionar un retraso en el lenguaje.
Se asocian también malformaciones cardiacas (25%) de grado variable, lo más frecuente son los defectos del tabique.
En relación con el crecimiento en general la talla es baja pero proporcionada.
Existen curvas de crecimiento de peso, talla y perímetro cefálico especiales para niños y niñas con SdCL con las que se puede seguir su crecimiento. En caso de observar una desviación respecto a estas curvas, se deben estudiar otras patologías asociadas.
No se deben dar suplementos calóricos que no mejoran el crecimiento salvo en caso de que existan criterios específicos.
Algunos individuos presentan defecto de hormona de crecimiento y requieren tratamiento exógeno. La hormona de crecimiento no mejora la talla final cuando no existe déficit de la misma.
La aparición y curso de la pubertad ocurre de forma similar a la población general, tanto en cuanto a cambios fisicos como en el campo emocional y del comportamiento.
La menstruación puede suponer un problema para los cuidadores, sobre todo en mujeres con problemas de comunicación. Además es importante prevenir embarazos desde el momento en que se inicia el periodo fértil. Existen diversas posibilidades terapéuticas de contracepción entre las que se elegirá la más adecuada para cada situación.
Entre los varones, está aumentada la frecuencia de testes no descendidos (casi un 10% frente a un 2% en la población general). El tratamiento médico tiene un alto índice de fracaso por lo que en general requieren descenso quirúrgico, para evitar riesgo de malignización en escroto.
Bibliografía
- http://ahedysia.org/patologias/116-síndrome-de-cornelia-de-lange
- De Lange CC: Sur un type nouveau de dégéneration (typus Amstelodamensis). Arch Med Enf Paris 1933;36:713-719.
- Fundación Cornelia de Lange USA. CdLSUSA /publicación periódica en línea/2006/Fecha de consulta 2006
- Jackson L y cols: de Lange Syndrome: a clínicasl Review of 310 Individuals. Am J Med Genet 1993;47:940-946.
- Opitz JM: The Brachmann-de Lange syndrome. Am J Med Genet 1985;22:89-102.
- Preus M, Rex AP: Definition and diagnosis of the Brachmann-de Lange syndrome. Am J Med Genet 1983; 16:301-312.
- Schaumann B, Alter M: Dermatoglyphics in Medical Disorders.Springer-Verlag. New Y.1976.Pag:196-199.
- Beck B, Fenger K: Mortalidad, hallazgos anatomopatológicos y causas de muerte en el síndrome de De Lange. Acta Paediatr Scand (ed esp) 1985;2:843-847.