INFORMACIÓN BÁSICA: Keratodermias.- Las keratodermias Palmoplantares (PPKs) son un grupo diverso de enfermedades de la piel caracterizadas por el hiperkeratosis palmoplantar de la epidermis. A la fecha, la mayoría de PPKs se ha atribuido a las mutaciones en las proteínas estructurales como las queratinas, las proteínas del desmosomal y conexiones. Las queratinas son reguladores de los filamentos intermedios (IFs) de keratinocitos y se deforma en un espectro de desórdenes epidérmicos. Los Desmosomas son péptidos de las uniones de célula-célula que se encuentran predominantemente en el corazón y la piel.
Ellos unen como redes a la membrana celular y funcionan en la arquitectura del tejido manteniendo la integridad estructural. Los Desmosomas consista en varias proteínas de membrana y son dos, la desmogleina 1 y desmoplakina. Sabemos que en la misma molécula, o sea, en la desmoplaquina, varios genes pueden estar comprometidos.
Estas onexinas son las proteínas mayores de uniones del hueco que son involucradas en la comunicación intercellular. A la fecha, mutaciones en dos conexinas, Cx26 y Cx31, han sido asociados con la enfermedad epidérmica. Interesantemente las mutaciones autosómicas dominantes en Cx26 están el síndrome de Vohwinkel con keratodermia asociada con la pérdida de la audición. Muchas otras alteraciones de PPK ocurren en síndromes asociados con el cáncer, el neuropatía, anormalidades de pelo e insuficiencia cardíaca. Como en la enfermedad epidérmica, las proteínas estructurales tienen previamente se implicado en la cardiomiopatía dilatada, con el envolvimiento, de distrofina documentada en la cardiomiopatía dilatada X-vinculada. Se piensa que la proteína distrofina liga la actina a los filamentos vía un N-término el dominio obligatorio. Las mutaciones en α・- el actin cardíaco él también se ha mostrado para causar la miocardiopatía dilatada. Otras proteínas estructurales asociadas con las cardiomiopatías dilatadas incluyen la laminina, la metavinculina, αα-dystroglycanα, y αα- αγ-sarcoglycan α.
El Síndrome de Carvajal es un proceso familiar y su forma de transmisión probablemente sea recesiva, concepto que descansa en el registro de una marcada historia de consanguinidad. Los padres fueron primos hermanos en 9 casos. Es una queratosis displásica, que puede asociarse a otras displasias mesodérmicas: miocardiopatía dilatada y signos de hiperelasticidad e hipermovilidad articular. Su histopatología es especial (imagen 502). En este nuevo síndrome se rompe el modelo simplista de hiperplasia total observada en otras queratodermias palmo-plantares genéticas familiares: hiperqueratosis ortoqueratósica, hipergranulosis, acantosis y papilomatosis. Aquí la hiperqueratosis epidermolítica es el principal suceso, al que se asocia una marcada disqueratosis y una gran espongiosis.
Precisamente Carvajal-Huerta describe tres familias de Ecuador que mostró la evidencia de herencia autosómica recesiva de un síndrome que afecta el pelo, piel y corazón. La enfermedad epidérmica radicaba en una alteración estructural de las PPK y la enfermedad del corazón era diagnosticado como una falla por miocardiopatía dilatada del ventrículo izquierdo. Todos los individuos afectados tenían manifestaciones clínicas de todos los tres desórdenes. El estudio genómico de estas tres familias, los miembros afectados de mostraron el gen mutado homozigosico a 6p23-p24 donde se encuentra la desmoplakina. Mutación que protege de la desmoplakina revelaron una sola tachadura del nucleotide en la cola dominio que lleva a un codon de la parada prematura y truncamiento detrás de dominio de la proteína.
Carvajal Huerta estudia y describe magistralmente un grupo de pacientes estudiados por muchos años, en su mayoría adolescentes que presentaban: queratodermias palmoplantares con pelo lanugo asociadas a malformaciones cardiovasculares e inclusive muerte súbita por miocardiopatía; sin duda alguna un síndrome cardiocutáneo. Esta nueva entidad aceptada por la comunidad médica mundial, describe una asociación clínico-histológica (posteriormente diría molecular) de corte genético no mencionado previamente en la literatura médica dermatológica mundial. El cuadro clínico presenta elementos constantes, antes descritos, además de otros elementos variables: queratosis psoriasiforme y vesículo-ampollosa; hipermovilidad e hiperelasticidad articular.
El pelo lanoso es el marcador clínico más importante del síndrome de Carvajal, al decir de su descubridor.
Ahora bien, como muchos de los elementos descritos por el propio Carvajal, coincidían con otra patología de similares características fenotípicas y considerado otro síndrome cardiocutaneo, me refiero al síndrome de la isla de Naxos, los hallazgos de secuencia genética demostraron mutaciones en el gen que codifica la plakloglobina, y no la desmoplaquina del síndrome de Carvajal.
Además, los pacientes portadores del síndrome de la Isla de Nexus, fallecen por miocardiopatía derecha (miocardiopatía arritmogena del ventrículo derecho).
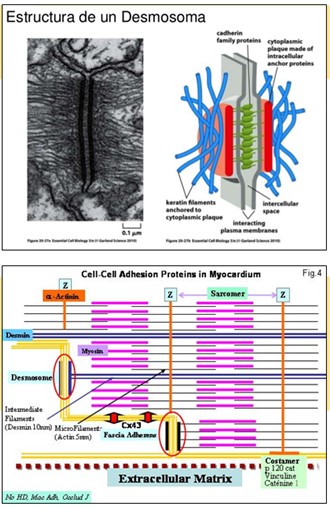
Sustrato anatomopatológico del Síndrome de Carvajal: Desmosomas: Los Desmosomas son los cruces principales de adhesión celular, particularmente prominente en la epidermis y el tejido cardiaco y son importantes para la rigidez y la resistencia de las células. El desmosome consta de varias proteínas, de las cuales desmoplaquina es el más abundante. Aquí, se describe la primera mutación recesiva humana, 7901delG, en el gen que provoca una desmoplaquina queratodermia estriada generalizada que afecta particularmente a la epidermis palmoplantar, pelo lanoso y una miocardiopatía ventricular izquierda dilatada. Varios de los pacientes con este trastorno sindrómica sufren insuficiencia cardíaca en sus años de adolescencia, lo que resulta en la morbilidad temprana. Todos los miembros afectados probados de tres familias de Ecuador eran homocigotos para esta mutación que produce un codón de parada prematuro dando lugar a una proteína truncada desmoplakina falta el dominio C de la región de la cola. Histología de la piel reveló grandes espacios intercelulares y la agrupación de los desmosomas en los sitios poco frecuentes de la adhesión de los queratinocitos. La inmunohistoquímica de la piel de los pacientes mostró una localización perinuclear de queratina en queratinocitos suprabasal, lo que sugiere una red de filamentos intermedios colapsado. Este estudio demuestra la importancia de la desmoplaquina en la unión de los filamentos intermedios de la desmosome. En contraste a null desmoplakina ratones que mueren en el desarrollo temprano, la proteína truncada debido a la mutación homocigótica 7901delG en los seres humanos no es letal embrionaria. Esto sugiere que no se requiere el dominio de cola de desmoplaquina para el establecimiento de la arquitectura del tejido durante el desarrollo.
Palmoplantar queratodermias (PPKS): Son un grupo diverso de enfermedades de la piel que se caracterizan por hiperqueratosis de la epidermis palmoplantar. Hasta la fecha, la mayoría de los PPKS se han atribuido a mutaciones en las proteínas estructurales tales como las queratinas, proteínas de los desmosomas, y conexinas. Las queratinas son las principales filamentos intermedios (FI) de los queratinocitos y se mutan en un espectro de trastornos epidérmicos. Desmosomas son uniones célula-célula se encuentran predominantemente en el corazón y la piel. Ellos enlace si las redes a la membrana celular y la función en el mantenimiento de la arquitectura del tejido y la integridad. Desmosomas consisten en un número de proteínas de membrana, dos de las cuales, desmogleína 1 y desmoplaquina, se ha demostrado que la base de autosómica PPK estriada no sindrómica dominante. Conexinas son las principales proteínas de las uniones intercelulares que están implicadas en la comunicación intercelular. Hasta la fecha, las mutaciones en dos conexinas, Cx26 y CX31, se han asociado con la enfermedad epidérmico. Curiosamente mutaciones autosómicas dominantes en Cx26 subyacen síndrome de Vohwinkel con la queratodermia mutilante asociado con sordera. Muchas otras formas de PPK se producen en los síndromes asociados con el cáncer (PPKS paraneoplásica), neuropatía, anormalidades de pelo y la insuficiencia cardíaca.
El síndrome de Vohwinkel es una genodermatosis de herencia autosómica dominante ocasionada por mutaciones en el gen GJB2, localizado en el locus 13q11-q12, que codifica para la conexina 26. Forma parte del grupo de las conexinopatías. Este seindrome se manifiesta desde la infancia y se hace más evidente en la edad adulta, y tiene predilección por el sexo femenino y la raza caucásica. Clínicamente, se caracteriza por una tríada: queratodermia palmo-plantar difusa, transgrediens, “en panal de abejas”; pápulas queratósicas lineales o “en estrella de mar” en codos y rodillas; y bandas fibrosas constrictivas en los dedos de manos y pies (pseudoainhum), a lo que se asocia hipoacusia moderada a grave.
Con la enfermedad epidérmica, proteínas estructurales previamente han sido implicadas en la miocardiopatía dilatada, con la participación de la distrofina documentado en la miocardiopatía dilatada ligada al cromosoma X. La proteína distrofina se cree que se unen los filamentos de actina a través de un dominio de unión a N-terminal. Las mutaciones en α-actina cardiaca en sí también se ha demostrado que causan la miocardiopatía dilatada. Otras proteínas estructurales asociados con miocardiopatías dilatadas incluyen laminina, metavinculina, α-dystroglycan, y α- y γ-sarcoglicano.
Tres familias de Ecuador que fueron estudiados mostraron evidencia de herencia autosómica recesiva de una enfermedad que afecta a sindrómica el cabello, la piel y el corazón. La enfermedad epidérmica era una forma estriada de PPK y la enfermedad cardiaca fue diagnosticado como un ventrículo izquierdo dilatado cardiomiopatía. Todos los individuos afectados tenían manifestaciones clínicas de los tres trastornos. Un genoma de exploración se lleva a cabo en estas tres familias, los miembros afectados de los cuales mostraron homocigosis en 6p23-p24, donde se encuentra desmoplakin. La detección de mutaciones de desmoplaquina reveló una única deleción de nucleótidos en el dominio de cola que conduce a un codón de parada prematuro y el truncamiento del dominio de la cola de la proteína.
SÍNDROMES CARDIOCUTÁNEOS: Síndrome de Carvajal-Huerta (OMIM 605676-615821/ORPHA:65282/CIE-10:I42.0/).- El Síndrome de Carvajal-Huerta (SCH) un síndrome cardiocutáneo que se caracteriza por queratodermia epidermolítica asociada a pelo lanoso, es un proceso que reúne condiciones clínico-histológicas y moleculares especiales. Básicamente se trata de un complejo queratodérmico, poliqueratósico, de probable transmisión recesiva, asociada a pelo lanoso, vesículo-ampollas y lesiones ungueales. Las alteraciones histológicas se caracterizan por constante presencia de hiperqueratosis epidermolítica, disqueratosis y espongiosis. En cuanto al pelo lanoso, es una distrofia del tallo piloso cuya textura está alterada al punto que el pelo sea fino y rizado.
Sinónimos.- Queratodermia con pelo lanoso tipo II (KWWH tipo II), Síndrome de pelo lanoso-queratodermia palmoplantar-miocardiopatía dilatada.
Prevalencia: <1 / 1 000 000
Epidemiología.- Sólo se han descrito unos pocos casos, todos ellos en pacientes procedentes de Ecuador, la India o Turquía.
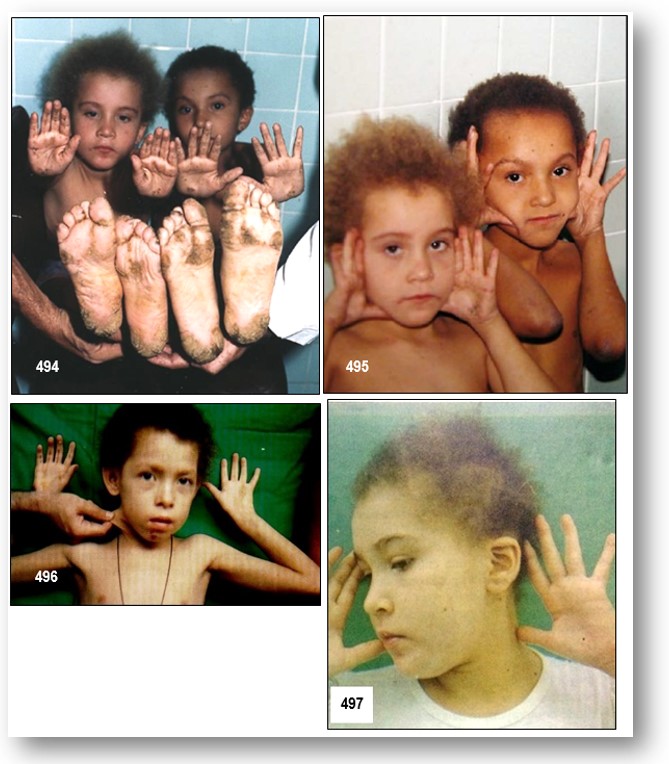
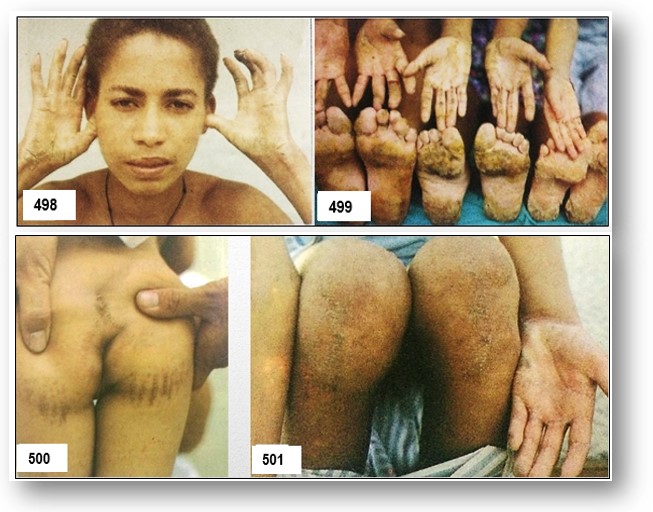
Características clínicas.- La enfermedad de la piel se presenta como un PPK con alguna participación no volar en particular en los lugares de presión o abrasión. Todas las lesiones epidérmicas tuvieron la hiperqueratosis longitudinal característica de un PPK estriado (Imagen 1 ). El cabello era rizado y lanoso en la textura. Investigación cardiológica incluyendo electrocardiograma y exámenes ecocardiográficos de varios de los miembros de la familia afectados se ha reportado con el diagnóstico de una miocardiopatía dilatada del ventrículo izquierdo a menudo resulta en insuficiencia cardíaca durante la adolescencia. La miocardiopatía dilatada se caracteriza por el agrandamiento cardíaco y la contracción cardíaca interrumpido.
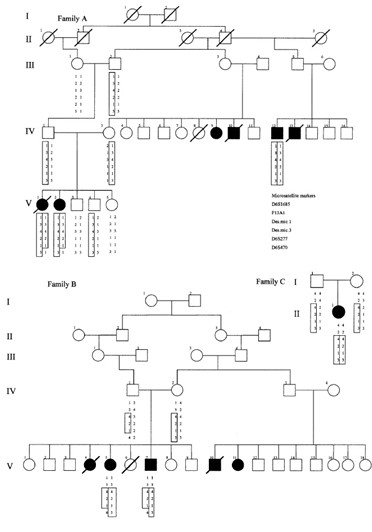
Defecto molecular.- Mapeo autozigótico.– El síndrome de Carvajal es causado por una mutación homocigótica en el gen que codifica para la desmoplaquina que trunca el extremo C de la proteína y se asigna al cromosoma 6p24. Puesto que no había pruebas de consanguinidad dentro de dos de las familias y las tres familias eran de la misma región del Ecuador, la hipótesis de que todos los miembros de la familia afectados eran propensos a haber heredado el mismo alelo de la enfermedad ancestral en los dos cromosomas maternos y paternos. Además, el síndrome es raro, ya que sólo había otra familia con manifestaciones clínicas similares ha sido reportado en la literatura. Hacia la identificación de la base genética de este síndrome, un genoma de exploración autozygosity se realizó con 386 loci de microsatélites marcadores para determinar el genotipo de todas las personas potencialmente informativos de los tres linajes de Ecuador.
El SCH se transmite como un rasgo autosómico recesivo y está causado por mutaciones en el gen DSP (6p24), que codifica para desmoplaquina, una proteína implicada en la adherencia celular.
La homocigosis en los individuos afectados se demostró con un marcador, F13A1, que se localiza en la región cromosómica 6p23-p24. Ninguno de los miembros de la familia no afectados eran homocigóticos para las enfermedades asociadas a F13A1 alelo segregar en cada familia.
Curiosamente, los miembros afectados de los dos pedigrees más grandes (A y B) fueron homocigotos para el mismo alelo, mientras que el individuo afectado del árbol genealógico más pequeña era homocigoto para un alelo diferente. Homocigosis para todos los individuos afectados no fue visto por cualquier otro marcador en el genoma de exploración. Para confirmar la vinculación y establecer si las tres familias habían heredado el mismo haplotipo ancestral enfermedad, marcadores adicionales de 6p23-p24 se genotipo a través de las genealogías. Además, los microsatélites fueron identificados a partir de la secuencia genómica de un cromosoma artificial bacteriano (BAC) clon (GenBank adhesión no. AL031058), que albergaba un gen candidato potencial para este trastorno, JUP (DSP). Microsatélites 1 (Des.mic.1) se encontró en el intrón 1 y 3 de microsatélites (Des.mic.3) en el intrón 23 del DSP de genes. Un análisis más detallado de haplotipos con estos marcadores de microsatélites vinculación adicionales confirmados y, efectivamente, revelan una segregación haplotipo enfermedad común en las tres familias (Imagen 55).
Desnaturalización cromatografía líquida de alto rendimiento (DHPLC) y análisis de secuencias
Desmoplakina mapas dentro del haplotipo enfermedad segregando en las tres familias ecuatorianas. Funcionalmente, desmoplaquina es un buen candidato para el gen de la enfermedad. Desmoplaquina es la proteína más abundante de desmosomas y es importante en la unión de la red si a la membrana plasmática y en la adhesión intercelular. Aunque las mutaciones haploinsuficiencia en DSP no resultan en la miocardiopatía o pelo lanoso, que están implicados en no sindrómica PPK estriada dominante en la que los portadores presentes con un patrón similar pero menos extensa de queratodermia. Las mutaciones en otras proteínas desmosómicas se han reportado para mostrar fenotipos epidérmicas similar a la segregación en las familias del Ecuador. Por ejemplo, una mutación en desmogleína 1 también resulta en autosómica estriada no sindrómica dominante PPK y los estudios de uniones desmosomales en el cabello sugieren que los defectos en las proteínas de los desmosomas, pueden producir una displasia hairshaft.
Se utilizó el ADN genómico de un afectado, uno no relacionada y un individuo heterocigoto para el haplotipo enfermedad para amplificar por PCR de cada uno de los 24 exones del DSP usando las condiciones descritas previamente. Los productos de PCR se sometieron después a heterodúplex formación y se analizaron usando DHPLC. En todos los individuos con genotipo como portadores, se observó un heterodúplex en el producto de PCR amplificado utilizando 24e cebador (Imagen 3 a). No se observaron más heterodúplex en los portadores en el resto del gen.
El análisis de secuencia del exón 24 reveló una deleción de un nucleótido G en la posición 7901 (7901delG con referencia a GenBank adhesión no.M77830) (Imagen 3 a) que segregó con la enfermedad en los tres familias con todos los miembros de la familia afectados homocigotos para esta secuencia variante. Este cambio no se observó en 100 controles normales no relacionados.
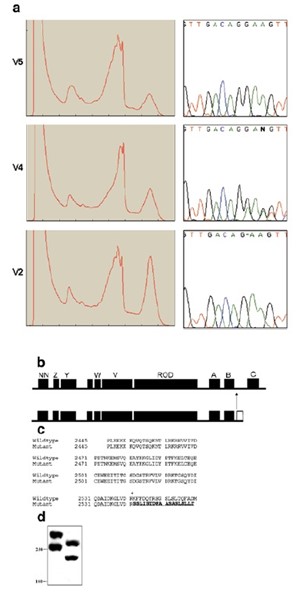
La mutación 7901delG, se predice para causar un codón de parada prematuro que se inserta 18 aminoácidos aguas abajo de la supresión y daría lugar a la truncación del dominio C en la región de la cola de la proteína (Imagen 503-3 b). Esta región de la proteína desmoplaquina se ha demostrado que interactúan con los fondos de inversión para anclar a la desmosome El análisis de transferencia de Western usando un anticuerpo anti-desmoplaquina confirmó el truncamiento predicha de la proteína desmoplaquina en queratinocitos cultivados de uno de los miembros de la familia afectados (Imagen 503-3 d).
Queda claro que la queratodermia epidermolítica asociada a pelo lanoso es un síndrome que puede ser integrada al grupo de las queratodermias palmo-plantares genéticas familiares en las cuales los engrosamientos palmo-plantares constituyen su marcador clínico.
El síndrome de Thiers-Chanial, es el único complejo queratodérmico conocido que muestra el mismo modelo del síndrome que describe Carvajal Huerta: en palmas queratodermia estriada y en plantas islotes callosos; pero se diferencia del síndrome de Thiers-Chanial por la ausencia de pelo lanoso y por presentar hipotriquia, onicogrifosis, hipoplasia dentaria y distrofias óseas. Lo que describe Carvajal-Huerta no se encontró en la literatura médica mundial, o sea, asociación de pelo lanoso, queratodermias palmar estriada con hiperqueratosis epidermolítica, disqueratosis y espongiosis. El pelo lanoso y la queratodermia palmar estriada por su constante presencia son los marcadores clínicos de este nuevo síndrome.
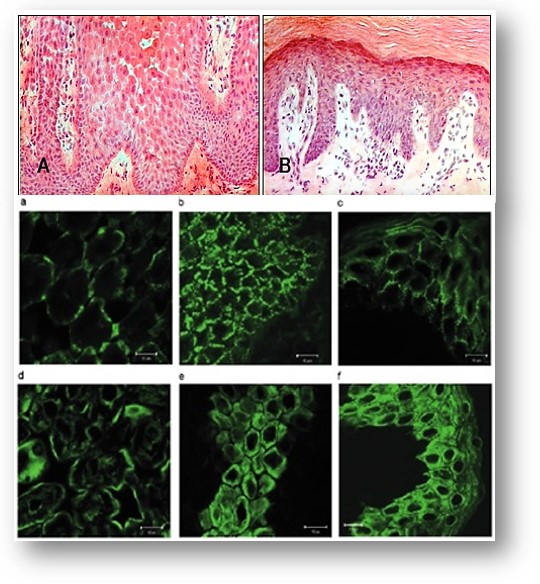
Histopatología investigación de epidermis.- Histología de la piel afectada paciente reveló grandes espacios intercelulares entre los queratinocitos suprabasal que apoyan el hallazgo de un defecto de adhesión celular que subyace a la enfermedad (Figura 502-A).Las células basales, sin embargo, parecían normales. La microscopía electrónica de palma afectada mostraba aglutinación de desmosomas en los sitios poco frecuentes de adhesión célula-célula (datos no mostrados).
La inmunohistoquímica sobre la piel afectada de palma.- Desmoplaquina tiene una distribución anormal en epidermis afectados en comparación con la observada en una biopsia de la palma de un individuo no relacionado no afectado. En queratinocitos suprabasal, desmoplaquina apareció principalmente en los puntos de contacto célula-célula (Figura 502-A). Otra proteína desmosomal, plakoglobina, mostró una redistribución similar (datos no mostrados). La queratina tipo II, KRT1, una proteína principal IF de queratinocitos suprabasal, ya no mostró una red de filamentos normal, pero tenía una localización membranosa y perinuclear (Figura 502-d). Queratinocitos basales suprabasal e inferior que no muestran los grandes espacios intercelulares tienen un desmoplakina aparentemente normal y distribución de queratina.
Diagnóstico diferencial del Síndrome de Naxos y el de Carvajal
– Enfermedad de Naxos: Ésta es una enfermedad muy rara, descubierta en la isla de Naxos (Grecia) y en otras islas Helénicas, en 24 pacientes de 6 familias. La forma de transmisión es recesiva; algunos otros casos aislados se han descubierto en el mundo.
- – Fenotipo: el fenotipo es idéntico a la Miocardiopatía arritmógena del ventrículo derecho (MAVD) clásica asociada con cabello lanoso y queratodermia.
- – Histología: típica asociación de MAVD. La asociación de signos de miocarditis y arritmias es frecuente, así como la muerte súbita.
- – Genotipo: la mutación del gen de la placoglobina (Proteína armadillo), es el factor monogénico que produce la enfermedad. Todos los pacientes son homocigotos. Los pacientes heterocigotos asintomáticos pueden tener taquicardia ventricular de Tracto de Salida del Ventrículo Derecho.
Enfermedad de Carvajal-Huerta: Ésta es una enfermedad muy rara, descubierta en Ecuador en 11 pacientes de 3 familias. Algunos otros casos aislados se han descubierto en el mundo.
- – Fenotipo: Muy similar al cuadro clínico del a isla de Naxos, pero con miocardiopatía arritmógena del ventrículo izquierdo (MAVI), asociada con cabello lanoso y queratodermias plamoplantares.
- – Histología: Signos de miocarditis.
- – Genotipo: Mutación del gen de la desmoplaquina (locus 6p24), factor monogénico que produce la enfermedad
- – Forma recesiva de transmisión, sindrómica, que afecta principalmente al ventrículo izquierdo, con compromiso del ventrículo izquierdo, los cardiomiocitos son sustituidos por tejido fibroso sin transformación adiposa, como sucede en el de la isla de Naxos.
- – Miocardiopatía arritmógena del ventrículo izquierdo, por desmoplaquina: controvertida.
Otras características del SCH incluyen queratosis folicular en codos y rodillas, cara, abdomen y extremidades inferiores, dedos en palillo de tambor, ampollas pruriginosas transitorias y queratosis psoriasiforme. La displasia se observa en el «síndrome similar a Naxos».
Conclusión: El SCH posee: Queratodermia palmoplantar epidermolítica, se trata pues de un síndrome poliqueratótico liderado por queratodermia palmar estriada, queratodermia plantar en “islotes” callosos, queratosis liquenoide estriada y queratosis folicular, Sólo en ciertos pacientes se forman vesículas y ampollas y queratosis psoriasiforme; pelo lanoso y disfunción ventricular izquierda inducida por miocardiopatía dilatada que terminan en muerte por paro cardiaco o en insuficiencia cardiaca. Desde el punto de vista molecular del trastorno, en el síndrome de Carvajal existe una mutación (cromosoma 6 p23-p24), una delección (Dsp7901del IG)) que codifica la desmoplakina, esta mutación causa una detención prematura la formación del codón, llevando a la formación de una desmoplakina truncada en el dominio C-Terminal. El SCH inicialmente había sido confundido con el síndrome de Naxos, tal como lo mencione en líneas anteriores, sin embargo, en esta entidad la clínica es muy similar, así como los cambios histopatológicos que se suceden, sin embargo la perturbación genética es diferente. En el síndrome de Naxos la mutación es una delección (Pk2157del2TG) que codifica la plakoglobina, esta mutación causa una terminación prematura de la traducción dando por resultado una proteína truncada en el dominio del C-Terminal. Estas mutaciones en ambas entidades afectan tanto a la piel como al miocardio, siendo más frecuente la afectación del ventrículo izquierdo en el SCH, y el ventrículo derecho en la Enfermedad de la isla de Naxos.
Comentarios de los Editores: La queratodermia epidermolítica asociada a pelo lanoso o SCH, es un proceso que reúne condiciones clínico-histológicas y moleculares especiales. Básicamente se trata de un complejo queratodérmico, poliqueratósico de probable transmisión recesiva, asociada a pelo lanoso, vesículo-ampollas y lesiones ungueales. Las alteraciones histológicas se caracterizan por constante presencia de hiperqueratosis epidermolítica, disqueratosis y espongiosis. El 50% de los pacientes desarrolla insuficiencia cardíaca y la mayoría de ellos muere durante la adolescencia si no se trasplantan. El gen alterado en esta enfermedad es el gen de la Desmoplaquina a (DSP), que codifica una proteína desmosómica: la DSP. En cuanto al pelo lanoso, es una distrofia del tallo piloso cuya textura está alterada al punto que el pelo sea fino y rizado. El patrón histológico, muy coherente por cierto, se caracteriza por una asociación especial de hiperqueratosis epidermolítica, disqueratosis benigna y espongiosis. El insólito descubrimiento y original síndrome genético familiar que más tarde llevaría honrosamente el nombre de unos de los médicos ecuatorianos más insignes del siglo XX. Los hallazgos descritos por Carvajal los hace conocer a la comunidad médica mundial por primera vez en 1998. El SCH reposa orgullosamente en la literatura médica mundial. Su descubrimiento fue el aporte de este hombre genial a la medicina ecuatoriana, del cual me enorgullezco haber sido su asistente clínico por más de 25 años.

Carvajal Huerta (f), mi Maestro; tuve la dicha y el honor de ser su asistente clínico en la Sala «Santa Luisa» y «San Pablo» del Servicio de Dermatología del Hospital General «Luis Vernaza Lasarte», Guayaquil-Ecuador, un sanatorio de más de 150 años, no solo el más antiguo sino también el más grande del país. La influencia de Carvajal sobre mi fue tan importante que motivó lo que soy actualmente, un curioso por descubrir en especial las patologías exóticas, en palabras académicas, soy investigador.
A mi maestro este humilde trabajo.

Bibliografía
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Derm. 39: 418-421, 1998.
- https://issuu.com/digitalmarketingnewyork/docs/sindrome_de_carvajal
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4830183/
- https://issuu.com/digitalmarketingnewyork/docs/sindrome_de_carvajal
- Norgett, E. E., Hatsell, S. J., Carvajal-Huerta, L., Ruiz Cabezas, J.-C., Common, J., Purkis, P. E., Whittock, N., Leigh, I. M., Stevens, H. P., Kelsell, D. P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Molec. Genet. 9: 2761-2766, 2000.
- Elizabeth E. Norgett, Sarah J. Hatsell, Luis Carvajal Huerta, Juan Carlos Ruiz Cabezas, John Común, Patricia E. Purkis, Neil Whittock, Irene M. Leigh, Howard P. Stevens, David P. Kelsell. Mutación recesiva en desmoplakina interrumpe desmoplakina-intermedios interacciones de filamento y causa la miocardiopatía dilatada, pelo lanoso y queratodermia. Human Molecular Genetic, Vol.9, N18, pag: 2761-2766
- Kelsell, DP y Stevens, HP ( 1999 ) Los queratodermias palmoplantar: mucho más que palmas y plantas. Mol. Med. Hoy en día , 5 , 107 -113.
- Corden, LD y McLean, WHI ( 1996 ) enfermedades de queratina Humanos:. Fragilidad hereditaria de los tejidos epiteliales específicos Exp. Dermatol. , 5 , 297-307.
- Rickman, L. et al. ( 1999 ) supresión N-terminal en una cadherina desmosómico hace que la autosómica dominante enfermedad de la piel estriada queratodermia palmoplantar. Hum. Mol. Genet. , 8 , 971 -976.
- Armstrong, D. et al. ( 1999 ) Haploinsuficiencia de desmoplakina provoca un subtipo estriada de queratodermia palmoplantar. Hum. Mol. Genet. , 8 , 143 -148.
- Maestrini, E. et al. ( 1999 ) Una mutación sin sentido en connexin26, D66H, hace que mutilar queratodermia con sordera neurosensorial (síndrome de Vohwinkel) en tres familias no relacionadas. Hum. Mol. Genet. , 8 , 1237 -1243.
- Richard, G. et al. ( 1998 ) Las mutaciones en el gen de la conexina GJB3 causa Eritroqueratodermia humano variabilis. Nature Genet. , 20 , 366 -369.
- Milasin, J. et al. ( 1996 ) una mutación puntual en el sitio de empalme 5 ‘del gen de la distrofina primera intrón responsable de X-linked miocardiopatía dilatada.Hum. Mol. Genet. , 5 , 73 -79.
- Olson, TM, Michels, VV, Thibodeau, SN, Tai, YS y Keating, MT ( 1998 ) mutaciones actina en la miocardiopatía dilatada, una forma hereditaria de la insuficiencia cardíaca. Ciencia , 280 , 750 -752.
- Towbin, JA ( 1998 ) El papel de las proteínas del citoesqueleto en las miocardiopatías. Curr. Opin. Celda. Biol. , 10 , 131 -139.
- Rao, BH, Reddy, IS y Chandra, KS ( 1996 ) ocurrencia familiar de una rara combinación de miocardiopatía dilatada queratodermia palmoplantar y con el pelo rizado. India Corazón J. , 48 , 161 -162.
- Whittock, NV y col. ( 1999 ) Estriado queratodermia palmoplantar resultante de desmoplakina haploinsuficiencia. J. Invertir. Dermatol. , 113 , 940 -946.
- Kurzen, H. et al. ( 1998 ) composición diferente desmosomas en los diferentes compartimentos del folículo piloso humano. Diferenciación , 63 , 295-304.
- Kouklis, PD, Hutton, E. y Fuchs, E. ( 1994 ) Establecimiento de una conexión: La unión directa entre los filamentos intermedios de queratina y proteínas de los desmosomas. J. Cell Biol. , 127 , 1049 -1060. Bornslaeger, EA, Corcoran, CM, Stappenbeck, TS y verde, KJ ( 1996 ) Rompiendo la conexión:. Desplazamiento de la proteína de los desmosomas placa desmoplakina de interfaces de célula-célula interrumpe el anclaje de haces de filamentos intermedios y altera conjunto de unión intercelular J. Cell Biol. , 134 , 985-1001.
- Smith, EA y Fuchs, E. ( 1998 ) La definición de las interacciones entre los filamentos intermedios y desmosomas. J. Cell Biol. , 141 , 1229 -1241.
- Gallicano, GI et al. ( 1998 ) JUP, se requiere al principio del desarrollo para el montaje de los desmosomas y la vinculación del citoesqueleto. J. Cell Biol. , 143 ,2009 -2022.
- Kowalczyk, AP et al. ( 1997 ) El dominio amino-terminal de desmoplaquina se une con plakoglobina y racimos desmosómicas complejos de cadherina-plakoglobina. J. Cell Biol. , 139 , 773 -784.
- Virata, ML, Wagner, RM, Parry, DA y Green, KJ ( 1992 ) La estructura molecular de la humana desmoplaquina I y II amino terminal. Proc. Natl Acad. Sci. EE.UU. , 89 ,544 -548.