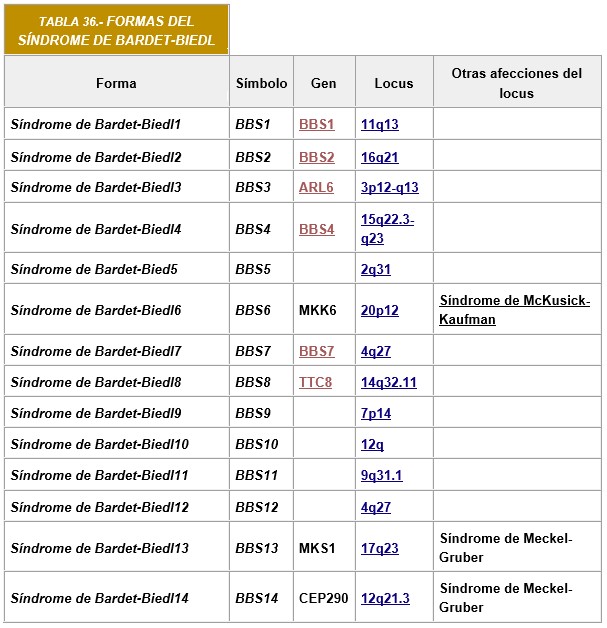
INFORMACIÓN BÁSICA: CILIOPATIAS: Síndrome de Bardet-Biedl (SBB) (OMIM 209900/ORPHA: 110). El SBB es una ciliopatía con afectación multisistémica. Su prevalencia en Europa está estimada entre 1/125.000 y 1/175.000. Existen no más de 200 casos descritos. Este trastorno está caracterizado por una combinación de síntomas clínicos: obesidad, retinopatía pigmentaria, polidactilia post-axial, riñones poliquísticos, hipogenitalismo y trastornos de aprendizaje, muchos de los cuales aparecen muchos años después de la aparición de la enfermedad. La expresión clínica es variable pero muchos de los pacientes manifiestan la mayoría de los síntomas clínicos durante el curso de enfermedad. La retinopatía pigmentaria es el único síntoma clínico constante después la infancia. El SBB puede también estar asociado con otras manifestaciones graves incluida diabetes, hipertensión, cardiopatía congénita y enfermedad de Hirschsprung (consulte este término). El amplio espectro clínico observado en el SBB está asociado a una significativa heterogeneidad genética. El trastorno se trasmite principalmente de manera autosómica recesiva pero se ha detectado herencia oligogénica en algunos casos. Hasta ahora, se han identificado mutaciones en 14 genes diferentes (BBS1, BBS12, MKS1, NPHP6/CEP290, SDCCAG8, and SEPT7 [septin 7]; véase la Tabla 1), también se asocia con mutaciones en el gen MKKS, que se localiza en el cromosoma 20, como responsables de los diferentes fenotipos del SBB. Estos genes codifican para proteínas SBB implicadas en el desarrollo y función de los cilios primarios. Los defectos en estos genes llevan a los cilios defectuosos. Estos cilios están implicados en el movimiento de la célula, en transmisión de señales químicas de los caminos de la entrada de información sensorial (tal como mirar, audiencia, y olor), etc.
La ausencia o disfunción de las proteínas SBB dan como resultado anomalías ciliares en órganos como el riñón y el ojo. Sin embargo, la relación entre los síntomas y la disfunción ciliar es desconocida para algunas de las manifestaciones clínicas del SBB. El reconocimiento del cuadro clínico es importante, ya que el diagnóstico puede confirmarse con análisis molecular, permitiendo el consejo genético apropiado para los miembros de la familia y el posible diagnóstico prenatal. El diagnóstico diferencial debe incluir los síndromes de Alström, McKusick-Kaufmann y Meckel-Gruber. Los pacientes con SBB necesitan una atención médica multidisciplinar. Las anomalías renales son la principal manifestación con riesgo para la vida porque pueden llevar a un fallo renal terminal que requiera trasplante renal. La pérdida progresiva de visión debido a la distrofia retiniana, junto con el déficit intelectual moderado (cuando está presente), anomalías en el comportamiento, hipomimia y obesidad afectan la vida social de estos pacientes.
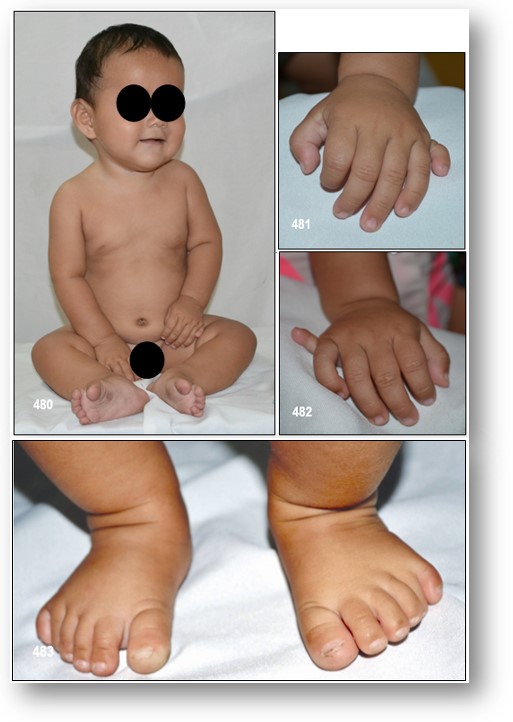
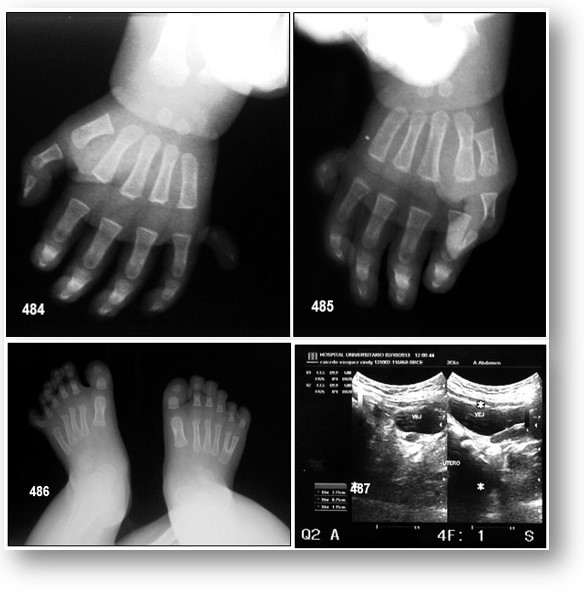
El SBB es un trastorno autosómico recesivo. A pesar de que los estudios genéticos han revelado 14 formas distintas del SBB relacionados con diferentes locus en distintos cromosomas el diagnóstico de SBB se sigue haciendo sobretodo en base a datos clínicos. Las manifestaciones clásicas incluyen: retinosis pigmentaria (RP), obesidad, polidactilia, hipogonadismo en los niños y retraso mental moderado. En general, los exámenes oftalmoscópicos y electrofisiológicos son esenciales para confirmar el diagnóstico de SBB. Las pruebas electrofisiológicas practicadas pueden demostrar potenciales evocados visuales (PEV) conservados, electrorretinograma (ERG) y amplitud disminuida, electrorretinograma (ERG) escotópico y fotópico abolidos. En un estudio realizado sobre 13 pacientes pediátricos Spaggiari et al, encontraron una disminución progresiva de la agudeza visual de manera precoz, en la primera década de la vida, signos de retinosis pigmentaria sólo en el 46% de los niños, grandes anomalías en el electrorretinograma de todos los pacientes, incluso en los que no presentaban RP, estos resultados electrorretinográficos, cuando se detectaron sugirieron mayor afectación del sistema fotópico. Como anomalías ortopédicas puede encontrarse polidactilia postaxial, escoliosis, tibia valga, tibia vara y otros trastornos, en nuestro caso sólo se apreció la polidactilia en manos y pies.
La expresión del SBB varía dentro de la misma familia y entre familias por lo que a menudo el diagnóstico es difícil. La edad media del diagnóstico estaba en 9 años, lo cual es tarde para esta afección debilitante, pero el lento desarrollo de los aspectos clínicos probablemente contribuye a ello. La polidactilia postaxial ya está presente en el 69% de los pacientes en el momento de nacer, pero la obesidad empieza a desarrollarse alrededor de 2-3 años y la degeneración retiniana no empieza a ser aparente hasta una edad media de 8,5 años. Como nuevos criterios clínicos se incluyen aspectos neurológicos, problemas del habla y lenguaje, alteraciones de conducta, dismorfismo facial y anomalías dentales. Incluyendo estos factores dentro de los criterios diagnósticos se podría facilitar un diagnóstico precoz de este trastorno. La evidencia de un fenotipo muy superpuesto al síndrome de Laurence-Moon ha llevado a una propuesta de unificación, con una descripción de síndrome polidactilia-obesidad y alteraciones reno- ocular.
Fisiopatogenia.- Diversas enfermedades monogénicas degenerativas como la enfermedad poliquística renal, la nefronoptisis, retinitis pigmentaria, el síndrome Bardet Biedl, el síndrome Joubert y el Meckel, pueden catalogarse como ciliopatías, un concepto reciente que describe las enfermedades que causan una disfunción de un organelo celular parecido al cabello llamado cilio.
Hay diferentes tipos de cilios, altamente conservados durante la evolución. Ellos participan en la transducción de una variedad de señales extracelulares que influyen en la polaridad, crecimiento neuronal, diferenciación o mantenimiento de los tejidos. Una amplia gama de señales puede recibirse por los receptores ciliares que incluyen la fotosensación, mecanosensación, osmosensación, termosensación, sensación hormonal y olfatoria.
Otros tipos de cilios móviles son estructuralmente similares a los cilios primarios, cuyas discinecias causan enfermedades genéticas como el síndrome Kartagener.
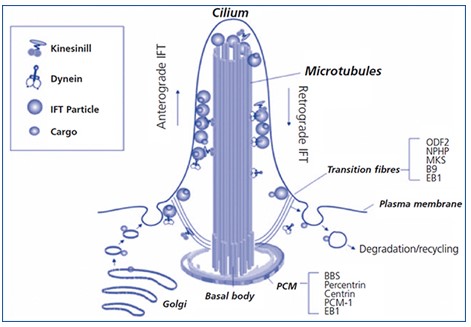
Pasare a revisar en detalles lo que es un cilio, su estructura y función del complejo de cilium-centrosoma (Figura 34). Los cilios (Et: del latín cilĭum, ceja, o tal vez del griego κυλίς, kilis, párpado o pestaña), son unos orgánulos exclusivos de las células eucariotas, que caracterizan por presentarse como apéndices con aspecto de pelo que contienen una estructura central altamente ordenada, constituida generalmente por más de 600 tipos de proteínas, envuelta por el citosol y la membrana plasmática. Algunos autores se refieren a las proteínas relacionadas con la función ciliar como «cilioma». Principalmente se trata de microtúbulos, que forman la parte central, llamada axonema. Aunque ya era ampliamente empleado en la literatura científica rusa de principios de siglo, Lynn Margulis propuso en 1985 el término undulipodio para referirse conjuntamente a los orgánulos que poseen estas características, los cilios y flagelos. La distinción entre éstos últimos se basa principalmente en su tamaño (unos 10-15 μm), número por célula (suelen ser muchos, con excepción de los cilios primarios y nodales, mientras que los flagelos uno o dos) y en su caso, por el patrón de movimiento (los cilios baten como un remo, son inmóviles o crean un vórtice, mientras que los flagelos ondulan).
Casi todos los eucariotas poseen células ciliadas, salvo los que tienen pared celular, que carecen habitualmente de ellos. Esto es especialmente cierto para los hongos y rodofíceas. En plantas existen las notables excepciones de algunos espermatozoides, como los de Ginkgo biloba o Cycas revoluta y los de criptógamas.
Los organismos aciliados tampoco poseen centriolos, por lo que algunos científicos creen que la función específica de éstos es la formación de cilios o flagelos. Significativamente, estos organismos tampoco poseen las tubulinas «especiales» (δ, ε, ζ y η) que permiten organizar el centriolo. En vertebrados, prácticamente todos los tipos celulares tienen cilios o proceden de células que los tuvieron. Los cilios móviles forman parte del epitelio del aparato respiratorio, del epéndimo o del aparato reproductor, mientras que los primarios se hallan virtualmente en cualquier tipo celular, como osteocitos, túbulo renal, fibroblastos y neuronas.
Dado su ubicuidad, están implicados en las funciones más diversas.
Los cilios móviles intervienen a la propulsión de organismos unicelulares, la limpieza de las vías respiratorias y el desplazamiento de los gametos, pero también contribuyen a regular el balance hídrico en los órganos excretores, la circulación de fluidos en la cavidad celómica, el sistema nervioso, el filtrado de partículas en las branquias. Los sensoriales contribuyen al reconocimiento de individuos compatibles en el apareamiento de protistas, mecanorrecepción en artrópodos, geotaxis en moluscos, reconocimiento y anclaje al hospedador en protistas parásitos y quimiorrecepción en vertebrados.
Así mismo existen muchas patologías derivadas de su mal funcionamiento, las denominadas «ciliopatías», como el síndrome de Kartagener, ciertos tipos de obesidad, el Síndrome de Laurence-Moon-Bardet-Biedl, el síndrome de von Hippel-Lindau o la enfermedad poliquística renal, entre otras, y también en algunos procesos de carcinogénesis. Algunos elementos celulares, como los estereocilios pueden confundirse con los cilios al microscopio óptico, pero en realidad están estructuralmente relacionados con las microvellosidades.
La enfermedad poliquística renal autosómica dominante y también la recesiva son cilipatías primarias, generadas por cambios en la poliquistina y policisteína que son importantes en la diferenciación de las células tubulares renales.
La nefronoptisis como causa de quistes renales corticomedulares, afecta primariamente los cilios por defectos de la nefroquistina e inversina.
La enfermedad de Von Hippel Lindau que causa hemangioblastomas múltiples es también una ciliopatía.
El síndrome Meckel con displasia renal, mielomeningocele, hipoplasia pulmonar, microftalmía y polidactilia; así como el síndrome Joubert con anomalías cerebelosas, discapacidad intelectual y colobomas retinianos, son también ciliopatías.
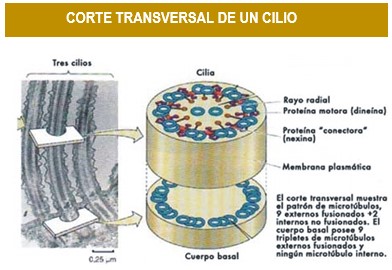
Cuatro mecanismos genéticos determinan el fenotipo en las ciliopatías:
- – Deleciones homocigóticas
- – Alelos múltiples
- – Genes modificadores
- – Oligogenicidad verdadera dos o más genes recesivos no son suficientes para expresar el fenotipo como es el caso de síndrome Bardet Biedl, donde se describen tres o más alelos mutados para causar el fenotipo.
Manifestaciones clínicas: Anomalías más características: Polidactilia (75-80%), distrofia de retina, obesidad (81-95%), retraso mental, retraso del crecimiento, nefropatías (incluida glomerulonefritis y riñones poliquísticos), sindactilia, hipoplasia genital, hipogonadismo hipogonadotrófico (desarrollo puberal incompleto y tardío suele ser frecuente). Según el hipogonadismo masculino o anormalidades genitales en mujeres se precisan cuando alcanzan la pubertad. Los varones con SBB, tienen genitales pequeños (pene y testículos). Las mujeres pueden presentar hipoplasia de trompas de Falopio, útero y ovarios, atresia total o parcial de vejiga, fístula vesicovaginal, himen imperforado, ausencia de orificio uretral, pueden experimentar ciclos menstruales irregulares. Las anormalidades estructurales y funcionales renales incluyen el doble sistema pielocalicial, divertículo de vejiga, hipoplasia renal, reflujo vesicouretral y fallo renal crónico.
Puede asociarse a diabetes insípida, fibrosis hepática, cardiopatías, nistagmus, retinitis pigmentosa (ceguera nocturna y la pérdida progresiva de la visión periférica), solo compromete al 15% de los casos. Estrabismo, clinodactilia del dedo meñique, hirsutismo, hiperplasia ovárica, enfermedad de Hirschsprung. Las funciones tiroidea, y suprarrenal suelen ser normales.
Otras anomalías clínicas: En algunos casos se han encontrado anomalías groseras, como atrofia del giro y lóbulos frontales, hidrocefalia, giro cerebral grande y distorsión de la simetría de los hemisferios cerebrales, ausencia de cuerpo calloso y adelgazamiento del mismo. Se hallaron algunas alteraciones microscópicas, como reducción del número de células, anomalías vasculares y gliosis. Véase la tabla 37.
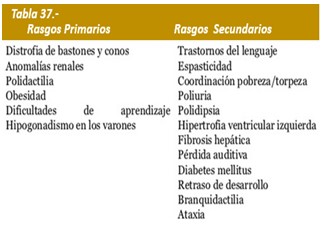
Se ha postulado que el SBB estaría asociado con hipopituitarismo o con alguna disfunción del diencéfalo o hipotalámica, esto no ha podido ser confirmado.
En un 70-85% existe retardo mental de distintos grados. Puede hacerse evidente precozmente o mas tardíamente con el transcurso de los años. En otros casos se ha descrito repentina pérdida de la función intelectual después de los 7 a 8 años de edad.
Puede existir: asociación con sordera, malformaciones renales, cardiopatías congénitas, malformaciones del cráneo y esqueleto (macrocefalia, braquicefalia, hidrocefalia, alteraciones en el tamaño de la silla turca, anomalías de vertebras y costillas, labio leporino y fisura palatina), atresia anal, hipertricosis, aspecto acromegálico) y mongoloides.
Alteraciones neurológicas y psiquiátricas: En raros casos se ha descrito asociación con esquizofrenia, psicosis paranoide, megalomanía, y dislexia.
Con mayor frecuencia pueden presentarse hipotonía e hiporreflexia, signos extrapiramidales, degeneración espinocerebelar, espasticidad e hiperreflexia y parálisis de nervios craneanos.
Se han observado frecuentemente anomalías electroencefalográficas tales como: actividad de fondo pobremente organizada y disritmias difusas. Las convulsiones son poco frecuentes.
Epidemiología: Esta condición es rara y se produce en todo el mundo con el cambio de frecuencias. Su tasa de prevalencia en Europa y América del Norte oscila entre 1:140.000 y 1:160.000, recién nacidos.
Por otro lado, es muy frecuente en población árabe Kuwait, así como en los Beduinos, y la isla de Terranova (Canadá), la tasa es muy alta, con un rango estimado de 1:17.000 nacidos vivos. También en población Árabe es generalmente alta, con una incidencia de 1 estimada en 17.000 nacidos vivos.
Diagnóstico.- El diagnóstico de esta condición no es muy fácil como el procedimiento de diagnóstico se basa en la presencia de 4 criterios primarios o tres primarios más dos secundarios.
La edad promedio para la detección del síndrome es de 9 años, cuando los problemas visuales comienzan a aparecer. Según el informe médico, sin embargo, hay casos en los que el diagnóstico se ha hecho después de 50 años de edad. También se dice que los médicos SBB pueden ser esencialmente sub-diagnosticada.
Las pruebas genéticas para SBB pueden confirmar el diagnóstico de este síndrome basa en la presencia de las características clínicas. Las pruebas genéticas con el fin de mutación en los genes SBB puede permitir el diagnóstico de la afección en los bebés que tienen polidactilia postaxial. Polidactilia se produce a una frecuencia de 1:1.500 en toda la población. En los bebés afroamericanos, las pruebas genéticas para SBB sólo pueden tener éxito si se padece polidactilia por otro síntoma de la BBS.
Diagnóstico molecular del síndrome de Bardet-Biedl mediante ARRAY BIOCORE: Este Array, contiene un panel de 129 mutaciones identificadas en los 13 genes más frecuentemente implicados en el diagnóstico de SBB:
Aproximadamente el 80% de los pacientes con el SBB tienen una mutación en alguno de estos genes. El ARRAY BioCore implica un gran avance ya que permite la identificación de 13 genes en un breve periodo de tiempo.
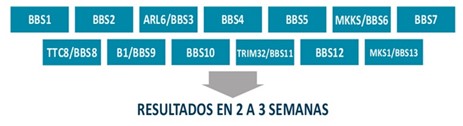
La aplicación de este panel contribuye a la identificación de las bases moleculares de las alteraciones clínicas del SBB.
NOTA IMPORTANTE: Este Array se ha desarrollado con el fin de mejorar la capacidad de diagnóstico el SBB. Sin embargo, un resultado negativo no excluye la posibilidad de que exista una mutación no incluida en este Array.
El dolor y la distensión abdominal constituyen uno de los motivos más frecuentes de consultas al médico (10% de las urgencias médicas). En general, se trata de procesos agudos autolimitados, pero cuando los síntomas se mantienen de forma crónica (prevalencia 25% de la población), pueden indicar la presencia de un número extenso de patologías, motivo por el cual el diagnóstico etiológico y diferencial es laborioso y costoso. BioCore ofrece un panel con las variantes genéticas más frecuentemente asociadas a trastornos gastrointestinales, que tiene como finalidad guiar en el diagnóstico diferencial de dichos trastornos.
Diagnóstico diferencial: Síndrome de Biemond tipo II, síndrome de Prader-Willi, síndrome de Cohen, síndrome de McKusick-Kaufmann (MKKS): se caracteriza por la tríada de hidrometrocolpus, polidactilia postaxial y cardiopatía congénita, su causa es por mutación del gen MKKS. El otro diferencial lo constituye el síndrome de Alström: retinosis pigmentaria, obesidad, sordera sensorial progresiva, cardiomiopatía dilatada, diabetes mellitus tipo dos y desarrollo motor tardío, a diferencia del SBB no hay deterioro cognitivo, ni polidactilia, tratándose de una mutación en el gen ALMS y se trasmite con carácter autosómico recesivo.
El estudio molecular no sustituye la evaluación clínica del paciente, considerándose un complemento valioso de la misma. El resultado debe evaluarse por un profesional con experiencia en consejo genético, por lo que se recomienda que el paciente analizado y su familia acudan a una consulta de genética clínica para la evaluación e interpretación precisa de los resultados.
El dolor y la distensión abdominal constituyen uno de los motivos más frecuentes de consultas al médico (10% de las urgencias médicas). En general, se trata de procesos agudos autolimitados, pero cuando los síntomas se mantienen de forma crónica (prevalencia 25% de la población), pueden indicar la presencia de un número extenso de patologías, motivo por el cual el diagnóstico etiológico y diferencial es laborioso y costoso. BioCore ofrece un panel con las variantes genéticas más frecuentemente asociadas a trastornos gastrointestinales, que tiene como finalidad guiar en el diagnóstico diferencial de dichos trastornos.
Diagnóstico diferencial: Síndrome de Biemond tipo II, síndrome de Prader-Willi, síndrome de Cohen, síndrome de McKusick-Kaufmann (MKKS): se caracteriza por la tríada de hidrometrocolpus, polidactilia postaxial y cardiopatía congénita, su causa es por mutación del gen MKKS. El otro diferencial lo constituye el síndrome de Alström: retinosis pigmentaria, obesidad, sordera sensorial progresiva, cardiomiopatía dilatada, diabetes mellitus tipo dos y desarrollo motor tardío, a diferencia del SBB no hay deterioro cognitivo, ni polidactilia, tratándose de una mutación en el gen ALMS y se trasmite con carácter autosómico recesivo.
Etiología y riesgo de repetición: Se han descrito hasta 5 loci implicados en la etiología de este síndrome. Se ha sugerido que los individuos heterocigotos portadores del gen mutado son propensos a la obesidad, hipertensión, diabetes mellitus y enfermedades renales. El riesgo de repetición es del 25% para hermanos de los afectados.
Procedimientos diagnósticos: Es posible detectar la polidactilia y las alteraciones renales por ecografía prenatal. Sin embargo, el diagnóstico es prácticamente imposible sin la existencia de un caso previo. Si existe una sospecha fundada, puede intentarse hacer el diagnóstico mediante análisis de ligamiento con procedimientos moleculares.
La continua investigación científica se dirige a localizar e identificar los genes que causan el SBB. Ya se han trazado los genes (localizando una región específica) en los cromosomas humanos (cromosoma 16). Esta investigación es el primer paso para el desarrollo de un medio de prevenir o tratar las características asociadas al SBB. El Síndrome de Laurence-Moon-Biedl-Bardet, no puede ser considerado como tal ya que en el SBB no existe paraplejía y en el Síndrome de Laurence-Moon no prevalece la polidactilia ni la obesidad y si existe parálisis. Por lo tanto son dos entidades diferentes.
Tratamiento: No hay ningún tratamiento para todas las características asociadas con el SBB. Definitivamente se tiene que controlar la obesidad con un plan de alimentación y educación en los hábitos alimentarios al entorno familiar desde edad temprana. La polidactilia se trata de manera quirúrgica. Cuando la visión empeora, los individuos se beneficiarán con del uso de ayudas de baja visión y entrenamiento en orientación y movilidad. Para manejar las complicaciones de la enfermedad renal asociada debe ser examinado por un nefrólogo. Tiene que considerarse terapia física, rehabilitación y según el retardo mental será el apoyo requerido.
Complicaciones: Algunas complicaciones se observan en pacientes que sufren de esta condición. Los individuos que sufren de SBB se sabe que tienen un mayor riesgo de desarrollar diabetes. Esto sucede debido a la obesidad o el defecto genético particular responsable del crecimiento de SBB. Todo ello conduce a un aumento de la glucosa en sangre que puede causar daño al corazón, los riñones, los ojos y los nervios. Las personas que sufren de SBB pueden tener problemas del corazón debido a la diabetes y el colesterol alto. Los problemas renales, que van de leves a graves, también pueden ocurrir. Si no se tratan adecuadamente, pueden ser potencialmente mortales en algunos casos.
Prevención: No hay prevención para SBB como tales. Sin embargo, el asesoramiento genético preconcepcional y genotipificación de los miembros de la familia pueden ser eficaces en la prevención de esta enfermedad. El tratamiento adecuado es obligatorio tan pronto como los síntomas sean evidentes. Las personas afectadas con SBB pueden vivir hasta 50 años.
Comentarios de los Editores: Los casos con este síndrome han sido incorrectamente incluidos en una entidad denominada síndrome de Laurence-Moon-Bardet-Biedl o síndrome de Laurence-Moon-Biedl, que no existe como tal. El síndrome de Laurence-Moon es una entidad distinta que no presenta obesidad ni polidactilia. La gran variabilidad, incluso entre hermanos, presente en este síndrome hace imposible su diagnóstico si los defectos presentes incluyen únicamente retraso mental e hipoplasia genital. La identificación de los genes involucrados en las diferentes formas genéticas de este síndrome podría ayudar a entender la etiopatogenia de algunos trastornos comunes de la edad adulta, como la obesidad, la hipertensión o la diabetes.
Bibliografía
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999; 36: 437-446.
- Lynn, Margulis; Heather I. McKhann, Lorraine Olendzenski (1993). «Introducción». Illustrated Glossary of Protoctista: Vocabulary of the Algae, Apicomplexa, Ciliates, Foraminifera, Microspora, Water Molds, Slime Molds, and the Other Protoctists (en inglés). pp. xviii. ISBN 0867200812.
- Spaggiari E, Salati R, Nicolini P, Borgatti R, Pozzoli U, Polenghi F. Evolution of ocular clínicasl and electrophysiological findins in pediatric Bardet-Biedl syndrome. Int Ophthalmol 1999; 23: 61-67.
- Ingster-Moati I, Rigaudiere F, Choltus-De Petigny MC, Bremond-Gignac D, Lestrade C, Grall Y. Functional visual explorations of Bardet-Biedl syndrome. A study of three cases. J Fr Ophtalmol 2000; 23: 802-808.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533-1543
- https://brainly.lat/tarea/428987