ACROCEFALOSINDACTILIAS
La acrocefalosindactilia (ACS) es una craneosinostosis sindrómica causada por mutaciones puntuales genéticas. Son frecuentes las alteraciones visuales y la deficiencia mental y pueden asociarse a una hipoacusia de conducción. La clasificación de la acrocefalosindactilia se realiza según la extensión y la gravedad de las anomalías craneales y de las extremidades. Algunos sistemas de clasificación comunes incluyen: Clasificación de Noordhoff y la clasificación de Cohen.
Hay varias enfermedades de origen genético relacionadas entre si:
Clasificación.-
-Acrocefalosindactilia
- – Acrocefalosindactilia tipo I o síndrome de Apert
- – Acrocefalosindactilia tipo III o síndrome de Saethre-Chotzen
- – Acrocefalosindactilia tipo V o síndrome de Pfeiffer
-Acrocefalopolisindactilia
- – Acrocefalopolidactilia tipo II o síndrome de Carpenter
- – Acrocefalopolidactilia tipo III o síndrome de Sakati-Nyhan-Tisdale
Se ha sugerido, por algunos estudiosos, que la distinción entre acrocefalosindactilia y acrocefalopolisindactilia debe ser abandonada.
Los síndromes de ACS representan un grupo de síndromes malformativos hereditarios congénitos caracterizados por craneosinostosis y fusión o unión de los dedos de las manos o los pies, asociada a menudo con otras manifestaciones.
Epidemiología.- La prevalencia exacta y la incidencia al nacimiento de los síndromes de ACS se desconocen. La incidencia global de todas las formas de craneosinostosis es de 1/2.000 a 2.500 nacidos vivos. Las cráneosinostosis sindrómicas ocupan un 40% de todas las craneosinostosis, en ese grupo, la acrocefalosindactilia tipo I ocupa un 4.5% y se presenta con una prevalencia de 1 caso en 65.000 nacimientos hasta 1 en 160.000 nacidos vivos, asimismo según el sexo, el Síndrome Apert puede aparecer en una proporción de 1 a 1 en hombres y mujeres.
Un estudio realizado en España en 1999 reportó resultados de hallazgos de una frecuencia del síndrome de 0,11 por 10.000 recién nacidos vivos, que es similar al descrito en la literatura internacional, con datos obtenidos del ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas) en el cual se tomaron en cuenta a 710.815 recién nacidos vivos entre 1976 y 1988.
Descripción clínica.- La acrocefalosindactilia incluye un número de síndromes con manifestaciones clínicas similares y a menudo solapantes. Todas ellas incluyen la craneosinostosis de una o múltiples suturas con rasgos faciales distintivos, déficit intelectual y del desarrollo variables, y diversas formas de anomalías de los pies y las manos. El grupo incluye los síndromes de Apert, Pfeiffer, Saethre-Chotzen, Jackson-Weiss y Carpenter.
Etiología.- Se han encontrado mutaciones en varios genes en los síndromes de ACS, incluyendo los siguientes: FGFR1 (8p11.23-p11.22), FGFR2 (10q25.3-q26), FGFR3 (4p16.3), y TWIST1 (7p21).
Consejo genético.- El patrón de herencia en los síndromes de acrocefalosindactilia es autosómico dominante (excepto en el síndrome de Carpenter, que es autosómico recesivo). Las mutaciones genéticas de novo son muy comunes, especialmente en los subtipos más graves. Se recomienda proporcionar asesoramiento genético.
INFORMACIÓN BÁSICA.- ACROCEFALOSINDACTILIAS: El Síndrome de Apert o Acrocefalosindactilia tipo 1 (OMIM 101200/ ORPHA:87). Pertenece a un grupo de cinco enfermedades caracterizadas, entre otras manifestaciones, por presentar craneosinostosis. Se suele clasificar como un síndrome del arco branquial, con afectación del primer arco branquial; que en los humanos es precursor del maxilar y mandíbula. Las perturbaciones en el desarrollo de los arcos branquiales en el desarrollo fetal provocan efectos duraderos y generalizados.
Las otras enfermedades que conforman el grupo son el síndrome de Carpenter, la enfermedad de Crouzon, el síndrome de Saethre-Chotzen y el síndrome de Pfeiffer. El síndrome de Apert es una enfermedad que puede ser hereditaria, o que puede también presentarse sin que existan antecedentes familiares conocidos. Esta condición se caracteriza por el cierre prematuro de las suturas craneales (suturas entre los huesos del cráneo), lo cual hace que la cabeza tome una forma puntiaguda y que se deforme la apariencia de la cara. El síndrome fue descrito, por primera vez, en el año 1906 por Eugene Charles Apert, un médico pediatra francés que le cedió su nombre. Numerosos estudios demuestran como el síndrome de Apert se produce hasta en un 98% de los casos por nuevas mutaciones que se generan en el periodo prenatal. Su herencia es de carácter autosómico dominante.
Fisiopatología.- Durante el desarrollo embriológico, las manos y pies se forman gracias a células que siguen un programa de muerte selectiva (apoptosis), lo que finalmente provoca la separación de los dígitos.
En los casos de acrocefalosindactilia, no se produce la muerte selectiva y por lo tanto la piel, y más raramente los huesos, de dedos de manos y pies permanecen unidos.
También existe una afectación de los huesos craneales, similares al síndrome de Crouzon o al síndrome de Pfeiffer.
La craneosinostosis ocurre cuando los huesos del cráneo y cara del feto se funden demasiado tempranamente, aún in útero, afectando el normal crecimiento de los huesos. La fusión de diferentes suturas conduce a diferentes patrones de crecimiento del cráneo. Las variantes incluyen: trigonocefalia (fusión de la sutura metópica), braquicefalia (fusión de la sutura coronal), dolicocefalia (fusión de la sutura sagital), plagiocefalia (fusión de las suturas coronal y lambdoidea en forma unilateral), y oxicefalia o turricefalia (fusión de las suturas coronal y lamboidal).
Algunos estudios relacionan la edad paterna elevada con los casos de novo del síndrome de Apert, e incluso han demostrado el origen paterno de las mutaciones.
Es importante dar un consejo genético a los que padecen la enfermedad, o a los que ya teniendo un hijo quieren saber qué posibilidades tienen de tener otro hijo afecto. Por lo general si los dos miembros de la pareja son sanos, y ya tienen un hijo con síndrome de Apert, existe un riesgo de aproximadamente un 1% de tener un nuevo hijo enfermo. En cambio, y debido a la modalidad de herencia de este síndrome, si una persona afectada quisiera tener un hijo existe un riesgo de un 50% de que pueda heredar dicha enfermedad.
Aunque los mecanisinos moleculares que condicionan el síndrome son complejos y no del todo conocidos, se han identificado diferentes mutaciones capaces de producir el síndrome, algunas de las cuales afectan al gen del «receptor del factor de crecimiento de los fibroblastos». Este receptor es fundamental a la hora de iniciar los mecanismos moleculares de señalización celular y, cuando es activado por sus ligandos, se produce una cascada de señales dentro de las células que son determinantes a la hora desencadenar y coordinar los mecanismos inlplicados en la fusión ósea. La alteración o no-regulación de estos mecanismos es la responsable final de la craneosinostosis.
La región extracelular de este receptor, que es la región que responde a los estímulos externos, está constituida por tres dominios IgI, IgII, lgIII (tipo inmunoglobulina).
Las mutaciones más importantes, descritas hasta la fecha, afectan a laregión de unión entre los dominios 11 y 111 y, en el axón 7 del gen para el «receptor del factor de crecimiento de los fibroblastos», se han descrito dos mutaciones claramente relacionadas con el síndrome; una mutación que condiciona la sustitución del aminoácido serina por triptofano, en la posición 252 (S252W), y otra que determina la sustitución de prolina por arginina (P253R).
El síndrome de Apert se caracteriza, de modo general, por el cierre prematuro de las suturas craneales, lo que hace que la cabeza tome una fomla puntiaguda y que se deforme la apariencia de la cara. Se producen malformaciones en cráneo, cara, manos y pies, además de diversas alteraciones funcionales que varían mucho de unos niños a otros.
Incidencia.– Estudios extensos muestran una incidencia de 1 y 6 casos por cada 100.000 nacidos vivos, sin preferencias de sexo.
Genética.- En base a la evidencia empírica la acrocefalosindactilia podría tratarse de un desorden autosómico dominante. Niños y niñas se encuentran igualmente afectados; sin embargo queda por investigar la causa exacta. Sin embargo, casi todos los casos son esporádicos, implicando mutaciones de novo o algún tipo de daño ambiental sobre el genoma. La descendencia de un padre con el síndrome de Apert tiene una probabilidad del 50% de heredar la enfermedad. En 1995, A.O.M. Wilkie publicó un documento que muestra evidencia de que acrocefalosindactilia se transmite de forma autosómica dominante, es decir, la presencia de una mutación del gen que codifica el Receptor 2 del Factor de Crecimiento de los Fibroblástico (FGFR 2) en el brazo largo del cromosoma 10:10q26, sin embargo no es el único síndrome relacionado con mutaciones de este gen, ya que, se sabe que otras alteraciones del mismo dan lugar a los síndromes de Crouzon, Pfieffer y Saethe-Chotzen. (Figura 33).
Puede haber dos tipos de mutaciones:
- – P253R. Responden mejor e la cirugía craneofacial pero tienen una sindactilia mas grave.
- – S252W
El síndrome de Apert es un desorden autosómico dominante; aproximadamente dos tercios de los casos son debidos a un cambio de una citosina por una guanina en la posición 755 en el gen FGFR2, lo que causa un cambio de una serina por un triptófano en la proteína. Este es un «punto caliente» para mutaciones específico de ascendencia masculina: en un estudio de 57 casos, la mutación ocurrió siempre en el alelo paterno del gen. En base a la prevalencia de la enfermedad entre los nacimientos (1 en 70.000), la tasa aparente de mutaciones C por G en este sitio es de aproximadamente 0,00005, lo cual es de 200 a 800 veces más alta que la tasa normal de mutaciones en dinucleótidos CG. Y aún más, la incidencia aumenta ajustadamente con la edad del padre. Goriely y colaboradores en el año 2003 analizaron la distribución alélica de mutaciones en las muestras de esperma provenientes de hombres con diferentes edades, concluyendo que la explicación más simple para los datos recaudados es que la mutación C por G provee a las células una ventaja en el desarrollo de la línea germinal masculina.
Todavía no se encuentra muy claro el porqué de que las personas que padecen el síndrome de Apert expresan ambas características, sindactilia y craneosinostonosis. Existe al menos un estudio que sugiere que esto se encuentra relacionado con la expresión de tres isoformas del gen FGFR2, el gen sobre el cual las mutaciones puntuales causan el síndrome en un 98% de los pacientes.
El receptor del factor de crecimiento de queratinocitos (KFGR), es la isoforma activa en la metáfisis y en las articulaciones interfalángicas. El FGFR1 es la isoforma activa en la diáfisis. El FGFR2-Bek es activo en la metáfisis y en la diáfisis, pero también en el mesénquima interdigital. La mutación puntual incrementa la activación dependiente de ligando del FGFR2, y también de sus isoformas. Esto significa que el FGFR2 pierde su especificidad, causando la unión del factor de crecimiento de fibroblastos (FGF) allí donde normalmente no debería unirse al receptor. Debido a que el FGF suprime la apoptosis, esto causa que el mesénquima interdigital se conserve. El FGF además aumenta la replicación y diferenciación de osteoblastos, causando por lo tanto la fusión temprana de varias suturas del cráneo.
Esto podría explicar porque ambos síntomas se encuentran siempre juntos en el síndrome de Apert.
Manifestaciones clínicas: El síndrome de Apert expresa sus signos y síntomas en varios niveles, los cuales incluyen: estructuras cráneofaciales, huesos de manos y pies, piel y aparato estomatológico.
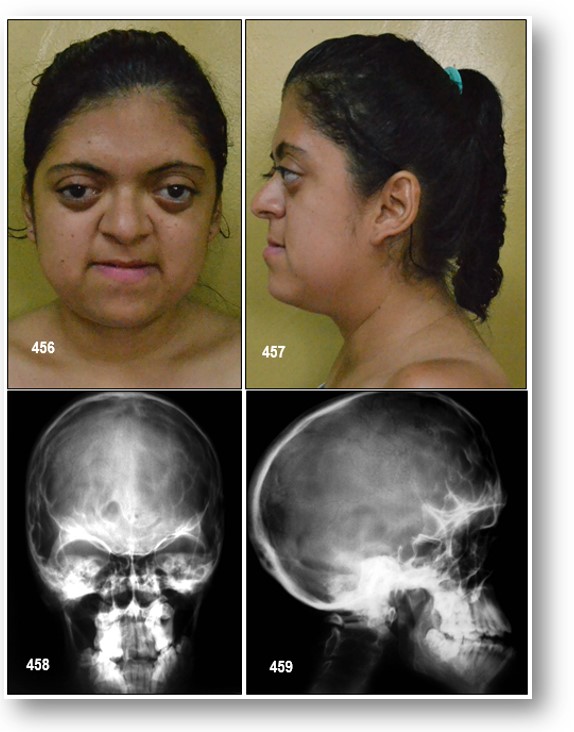
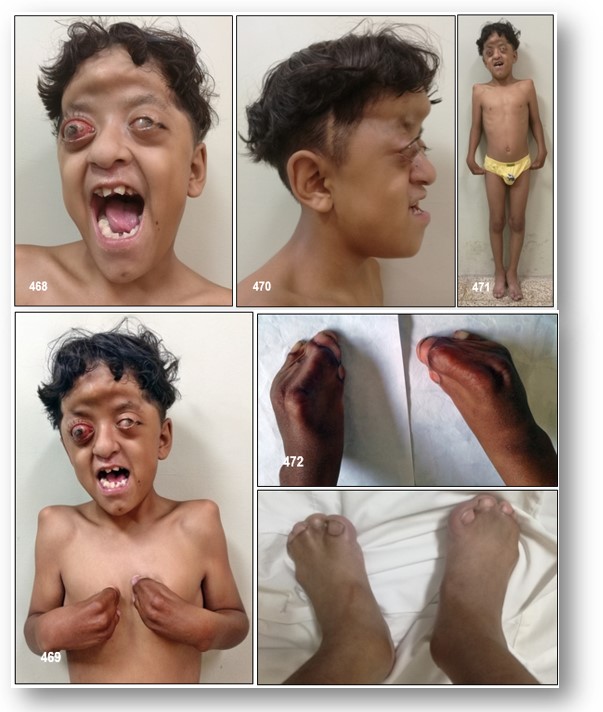
Alteraciones cráneo-faciales: Ya he mencionado que en el síndrome de Apert se produce una craneosinostosis como consecuencia del cierre precoz de las suturas del cráneo, desencadenando esto una deformidad de la bóveda craneal que pudiera impedir una expansión cerebral adecuada, y que se puede manifestar clínicamente en forma de edema papilar, atrofia óptica, e incluso hipertensión intracraneal. Los individuos con este síndrome presentan una hipoplasia, o escasa formación de la mitad de la cara, acompañada de ojos protruyentes (proptosis y exoftalmos), debido a una disminución del tamaño de la cavidad orbitaria. Los afectados pueden presentar macroglosia, maloclusión mandibular y en ocasiones asocian paladar ojival y fisura palatina.
Estas alteraciones morfológicas condicionan la aparición de infecciones frecuentes tipo otitis y episodios de dificultad respiratoria.
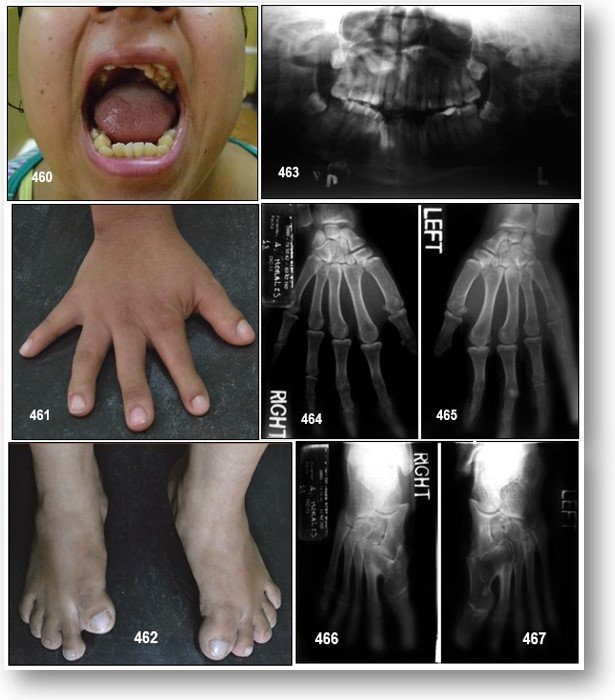
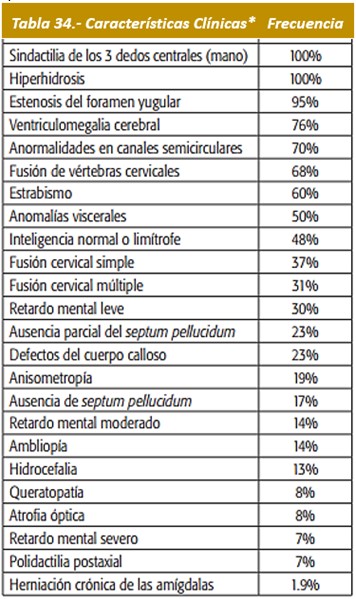
Manifestaciones dentales: Entre las características comunes pertinentes de acrocefalosindactilia se encuentran un paladar de arco alto, prognatismo pseudomandibular (que aparece como prognatismo mandibular), un paladar estrecho, y el apiñamiento desordenado de los dientes.
Hay presencia de mordida abierta, paladar ojival y, en ocasiones, fisurado, con úvula bífida. El paladar es corto, con una forma peculiar producida por engrosamiento fibroso por palatino de los procesos alveolares.
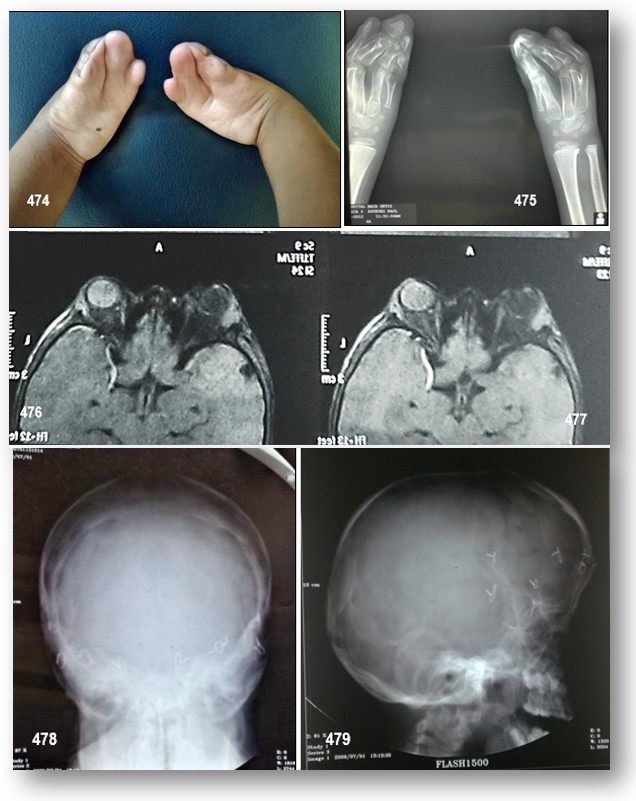
Manifestaciones músculo-esqueléticas: Característicamente estas personas presentan sindactilias, o fusión anormal de los dedos, en las cuatro extremidades, siendo más severas las que afectan a las superiores ya que en los dedos de los pies habitualmente no se fusionan las falanges distales. En función del número de dedos afectados se clasifican en:
- -TIPO 1: Incluye el 2′, 3″ y 4″ dedo.
- -TIPO 2: Asocia el 5″ dedo.
- -TIPO 3: Todos los dedos aparecen unidos.
Las malformaciones en el espacio entre el índice y el pulgar son variables. Basados en este primer espacio interdigital, es posible diferenciar tres tipos diferentes de malformaciones:
Tipo I: también llamada «mano espada». Que es el tipo más común y menos severo de malformación. El pulgar presenta una desviación radial y clinodactilia, pero se encuentra separado del dedo índice. El índice, medio y anular se encuentran fundidos en la articulación interfalangeal distal produciendo una palma plana. Durante el estadio embrional la fusión no tiene efecto sobre el crecimiento longitudinal de estos dedos, por lo que tienen una longitud normal. En el cuarto espacio interdigital siempre se observa una sindactilia simple, ya sea completa o incompleta.
Tipo II: también llamada mano de cuchara o en manopla. Esta es una malformación más seria debido a que el pulgar se encuentra unido al dedo índice por una sindactilia simple ya sea completa o incompleta, únicamente la falange distal del pulgar no se encuentra unida a nivel óseo con el índice y posee una uña separada. Debido a la unión de los dedos al nivel de las articulaciones interfalángicas distales, se produce una mano cóncava. La mayoría de las veces aparece también una sindactilia completa del cuarto espacio.
Tipo III: También llamada mano en «pezuña» o en «capullo de rosa». Esta es la forma menos común, pero también la más severa de las malformaciones en el síndrome de Apert. En esta existe una fusión sólida, ósea o cartilaginosa, de todos los dedos, con una única uña continua. El pulgar se encuentra girado hacia dentro y por lo tanto resulta imposible distinguir a cada dedo por separado. Por lo general resulta muy difícil obtener imágenes radiográficas o de resonancia de la mano debido a la superposición de los huesos, pero la exploración física por si sola no es suficiente para evaluar la gravedad de la malformación.
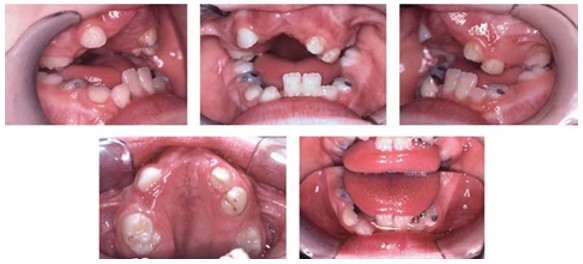
Imágenes 52 con fines Didácticos: Intraoralmente: Dentitio tarda, el hacinamiento de Angle Clase III con compensación dental por retrusión de los incisivos inferiores, mordida abierta circular con mordida cruzada unilateral. Fuente:
Esto es válido tanto para las manos, en las que predomina el tipo 1, como para los pies, que presentan con mayor frecuencia el tipo III. Además se ha demostrado como una de las mutaciones de este síndrome, la P253R, se asocia con más asiduidad a sindactilias más severas. Otras anomalías óseas menos frecuentes son: la fisión de vértebras cervicales, movilidad articular limitada, acortamiento de huesos largos como el fémur, húmero y el radio, hipoplasia de escápula, de pelvis … etc. En cuanto a la motricidad, pueden sufrir torpeza motora fina variable en función de la severidad de la sindactilia y los resultados conseguidos por la cirugía. El desarrollo motor grueso es normal, aunque con limitaciones por las características de los pies.
Manifestaciones dermatológicas: Son muy variables en cuanto a su expresividad y puede existir una hiperhidrosis, acompañada de lesiones máculo-vesiculosas o costrosas tipo acneiforme con propagación desusada a los antebrazos. En ocasiones presentan áreas de hipopigmentación (albinismo) y engrosamiento de la piel. A continuación describo en detalles las diferentes alteraciones dermatológicas:
1- Hiperhidrosis
Es el resultado de la disfunción de las glándulas apócrifas. En un estudio de 136 pacientes se la pudo constatar en el 100% de los casos; este aumento de la sudoración suele ser más ostensible en axilas, con una característica que la hace más desagradable, el mal olor (bromhidrosis). Los intentos infructuosos para atenuarla han incluido en el pasado hasta técnicas quirúrgicas.
2- Lesiones acneiformes
Solomon las describió primero en el año 197; otros han notado la persistencia de sus brotes, algunos muy severos y resistentes a los tratamientos habituales. Recientemente se obtuvieron buenas respuestas con isotretinoína y por separado con anticonceptivos orales.
La historia natural de esta anomalía comienza en la adolescencia o tiempo después, cuando la piel comienza a tornarse oleosa, para luego aparecer pápulas foliculares similares al acné, pero con una diferencia respecto del «clásico»: las pápulas no solo se ubican en cara y tronco sino también en antebrazos, glúteos y muslos, incluso se las ha observado adoptando una distribución «nevoide».
Ciertas mutaciones somáticas del Factor de Crecimiento de los Queratinocitos (RFCQ) determina un mosaicismo epidérmico, el cual provocaría anomalías del aparato pilosebáceo, o un aumento de la sensibilidad a los andrógenos de la unidad folículo-sebácea, o ambos fenómenos combinados, cuyo producto final es el brote acneiforme.
3- Sinoniquias
Esta alteración suele afectar más a las manos que a los pies y la asociación de falanges distales con sinoniquia, puede ser hallada en las manos pero nunca en los pies.
4- Uñas quebradizas
La causa de esta anomalía en el SA no ha sido establecida todavía, se sabe además que existen otras enfermedades, congénitas o no, que la exhiben. Dentro de las congénitas se cuentan el Síndrome de Hutchinson Gilford y la displasia ectodérmica con ectrodactilia.
5- Interrupción del trazado normal de las cejas
Está motivado por un defecto óseo subyacente, el cierre precoz de la sutura esfenoparietal, lo que crea una lámina orbitaria del frontal muy corta; esto provoca a su vez una gran retracción y elevación supraorbitaría, más pronunciada lateralmente, resultando en una solución de continuidad del trazado normal de las cejas.
6- Hipopigmentación de la piel, pelo y ojos (albinismo óculo-cutáneo)
Casi el 27% de los pacientes con SA presentan hipopigmentación de la piel, pelo y ojos. De nueve pacientes con SA observados por Margolis y col cinco mostraban esta anomalía. El color del pelo variaba desde castaño claro a rubio, la piel era pálida y los írides eran verdosos o azulados, habiéndose demostrado que la causa de esta hipopigmentación es la falta de migración de los melanoblastos in útero. Se ha observado además transiluminación del iris e hipopigmentación del fundus, asociado con ausencia o disminución de los reflejos foveales. No se demostró nistagmo pendular ni disminución de la agudeza visual, diferente a lo que sucede en la mayoría de las formas del albinismo óculo-cutáneo.
Las pruebas indican que la pérdida de la pigmentación y las anomalías esqueléticas en el SA son el resultado de alteraciones independientes, genéticamente relacionadas, de un proceso sucedido en un punto común, durante la gestación.
7- Hiperqueratosis plantar lateral
Esta es debida a la fusión ósea progresiva de los huesos del tarso y metatarso, lo que ocasiona transferencia del peso corporal hacia la región media y lateral del pie; el acto de caminar y estar de pie provoca luego el aumento de espesor de la piel.
8- Perionixis
Se observa con mayor frecuencia en pacientes institucionalizados y suele localizarse más habitualmente en las uñas de los pies que en las manos. La colonización periungueal por cándidas estaría favorecida por la existencia de sinoniquia, uñas quebradizas e hiperhidrosis.
9- Arrugas excesivas en la piel que cubre la frente
Consecuencia de la hipoplasia ósea del hueso frontal, la frente en el SA suele ser abultada e inclinada; dicha arquitectura y la ley de la gravedad, serian las responsables del exagerado arrugamiento de la región frontal (Cohen y col, 1995).
10- Hoyuelos (depresiones o fositas) en los nudillos, hombros y codos
Pueden aparecer desde la infancia, siendo esta localización poco frecuente; su localización es más típica en cara o salientes óseas, aunque se han observado fositas escapulares supraespinosas congénitas aisladas o asociadas al síndrome de la Trisomía 9p, o el Síndrome Russell-Silver, o en la delección del cromosoma 18q o en el Síndrome del pterigión poplíteo y en el Síndrome de los hoyuelos autosómicos dominantes. Es necesario destacar que en algunas infecciones tales como la rubéola congénita, en algunos traumas mecánicos y más raramente en formas de hipofosfatemias adquirida, se pueden observar estas fositas.
Manifestaciones viscerales: Aunque estas personas presentan, con frecuencia, un retraso mental y psicomotor secundario a sus anomalías morfológicas, ocasionalmente asocian malformaciones del sistema nervioso central que afectan al cuerpo calloso (agenesia o hipoplasia del mismo), y a las estructuras líbicas. También puede existir una hipoplasia de la sustancia blanca y otras.
Las alteraciones génito-urinarias son también frecuentes. En varones pueden aparecer válvulas uretrales posteriores que, factiblemente, dan lugar a hidronefrosis secundaria e insuficiencia renal si no se solucionan con prontitud y, en las hembras, pueden presentarse casos de hipertrofia de clítoris.
Las malformaciones cardíacas como la hipoplasia ventricular izquierda, la comunicación interauricular y la coartación de aorta, son manifestaciones típicas en las personas afectadas.
Características psicológicas .- No existen estudios rigurosos que confirmen la existencia de una configuración de rasgos de personalidad específica en las personas con síndrome de Apert, más allá de las descripciones de las historias de vida.
En general, las manifestaciones-psicológicas de la persona, su peculiar forma de ver el mundo, su afectividad y manejo de emociones y sus disposicioiies y tendencias a la acción, van a estar mediatizadas, por una parte, por la severidad de las manifestaciones clínicas anteriormente descritas y, por otro, por la existencia y, en su caso, intensidad del retraso mental. En los niños sin retraso mental son frecuentes los sentimientos de baja autoestima y miedo al rechazo, por ser diferentes o por sus deformidades físicas; dichos sentimientos pueden estar atenuados en los niños con retraso mental asociado al síndrome. Otros factores que van a influir en las repercusiones psicológicas del síndrome son, indudablemente, el apoyo social y la actitud del entorno y de las figuras emocionalmente significativas para la persona.
En el área del lenguaje pueden observarse dificultades de articulación debido a la hipoplasia del tercio medio de la cara, sobre todo en palabras que terminan en consonante. El lenguaje expresivo es pobre aunque, por lo general, expresan emociones sin dificultad. Es frecuente detectar dificultades de atención significativas.
Exploración semiológica: Al examen físico se puede encontrar:
Craneo y cara: Diámetro craneal AP disminuido, frente alta, craneosinostosis irregular (desuturacoronal) confontanelas grandes, cara plana, estrabismo, tendencia aproptosis de ojos, hipertelorismo, nariz pequeña, hipoplasia maxilar, paladar estrecho, úvula bífida, alteraciones dentales. Implantación baja de las orejas.
Extremidades: Sindactilia ósea o cutánea, normalmente con fusión del 2, 3 y 4 dedos de la mano, o toda la mano, falanges distales de los pulgares anchas y dedos cortos. Sistema nervioso central: Agenecia de cuerpo calloso, ventrículo megalia no progresiva, hidrocéfalo progresivo (con hipertensión endocraneana), alteraciones del hipocampo. Piel: En la adolescencia aparece acné de moderado a intenso e incluye los antebrazos; hiperhidrosis, microniquia y albinismo.
Otras alteraciones: Fusión de vertebras cervicales, habitualmente C5 y C6. Malformaciones cardiacas y gastrointestinales, aplasia pulmonar, alteración del cartílago traqueal, riñones poliquísticos, humero corto, criptorquidia, anquilosis decodos hombros y/o cadera. Otra característica clínica es la presencia de síntomas, como es la dificultad para la fonación y timbre nasal.
Crecimiento. Media de altura y peso
Funcionalidad. El retraso mental está presente en un número significativo de pacientes, según estudios el CI medio fue de 74.
Diagnóstico prenatal: El estudio prenatal se debe realizar en aquellas personas en las que exista un antecedente de la enfermedad o sospecha de la misma. Las ecografias realizadas durante el embarazo nos muestran signos inespecíficos que nos obligan a completar el estudio. Pueden aparecer: malformaciones de la bóveda craneal, ventrículomegalia cerebral, a expensas de los ventrículos laterales, sindactilia de las manos y pies y polihidramnios. Por lo expuesto, el estudio molecular y genético se realiza habitualmente en el primer o segundo trimestre de la gestación mediante la obtención de muestras por amniocentesis o por biopsia de vellosidades coriónicas y procesándolas mediante técnicas moleculares de reacción en cadena de la polimerasa (PCR).
Estudio postnatal: El estudio postnatal definitivo también se basa en la detección de mutaciones específicas. Es necesaria la realización de radiografías de cráneo, manos y pies que muestren las afectaciones óseas. Aunque las malformaciones del sistema nervioso central no son muy frecuentes, es asimismo necesario realizar una resonancia magnética para descartarlas. Se realizarán también estudios auditivos y visuales además de un estrecho seguimiento.
Diagnóstico: Se puede confirmar mediante una radiografía de cráneo y la exploración clínicas. En los niños con síndrome de Apert se requiere, en primer lugar, identificar un posible retraso mental a través del diagnóstico con la menor demora posible. Este diagnóstico permitirá determinar la intensidad de los apoyos en su rendimiento en el área de las habilidades adaptativas. Una adecuada evaluación de estas habilidades requerirá numerosas observaciones en distintos contextos, un juicio clínico y un análisis de las intensidades iniciales de apoyos para una persona, así como una nueva evaluación transcurrido el tiempo.
Resulta esencial seguir las recomendaciones de la AAMR en cuanto a eliminar la utilización de etiquetas del nivel de severidad para determinar un emplazamiento educativo.
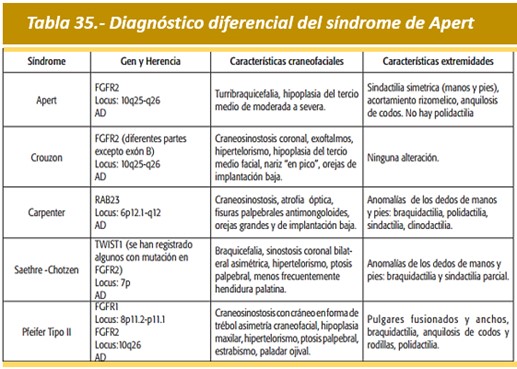
Diagnóstico diferencial.- Es necesario establecer diagnóstico diferencial del Síndrome de Apert con otros síndromes que presentan craneosinostosis (Tabla 35). Incluyen: Síndrome de Pfeifer, Síndrome de Crouzon, Síndrome de Carpenter, síndrome de Summit, el síndrome de Seathre-Chotzen y otros varios síndromes de craneosinostosis. En el síndrome de criptoftalmos puede observarse sindactilia acentuada, que se asemeja en la observada en el síndrome de Apert.
Diagnóstico Radiológico.- Radiográficamente hay huellas digitiformes craneales, coanas estrechas y paladar arqueado entre otras alteraciones óseas.
Análisis de las tomografías computadas (TAC) de cerebro de individuos con esta patología muestran un defecto en la maduración del cartílago de la base craneana que lleva a una hipoplasia mediofacial. Las suturas coronales frecuentemente están involucradas; las suturas lambdoideas no suelen estar comprometidas, lo que permite diferenciarlo del síndrome de Crouzon. Por otro lado, la cabeza suele ser alta y estar acortada en el sentido anteroposterior (braquicefalia). Otros hallazgos clínicos incluyen sindactilia de pies y manos, malformaciones en la articulación del codo y malformaciones cerebrales como agenesia del cuerpo calloso y malformaciones en el septum pellucidum, por lo que se produce retraso mental.
Tratamiento: Ámbito médico. El manejo de las personas que padecen este síndrome se basa en la corrección de las malformaciones morfológicas y en el apoyo psicológico a los enfermos. La cirugía correctora se debe realizar lo más precozmente posible y por múltiples especialistas.
Los neurocirujanos se encargan de reconstruir la bóveda craneal, ya que disminuyendo la presión intracraneal se evita la hidrocefalia y se favorece el desarrollo normal de las estructuras cerebrales; también es posible aumentar el volumen de las cavidades orbitarias.
Los especialistas en cirugía maxilofacial se encargan de las malformaciones de la cara. Esta intervención consiste en la realización de un adelantamiento de la mitad de la cara hipoplásica, lo que supone una intervención muy importante.
Habitualmente se realiza entre los 4 y 6 años de edad con el fin de evitar el impacto psicológico que suponen estas malformaciones durante la pubertad. La transformación de la cara requiere varias intervenciones por regiones y consisten en realizar osteotomias de corrección que incluyen el frontal y ambas mandíbulas. Otros especialistas, como odontólogos y ortodoncistas, también contribuyen a la reparación y reconstrucción de la zona oral.
Las sindactilias de manos y pies también precisan de una intervención quirúrgica, siendo esta realizada por los traumatólogos-ortopedas. La cirugía de la mano tiene como objetivo conseguir una correcta función de la misma. En un primer momento lo más importante es conseguir un espacio adecuado simulando la palma de la mano mediante una plastia en forma de «Z» y que el primer dedo realice correctamente el movimiento de oposición para que pueda agarrar objetos. Posteriormente se va remodelando la mano y separando los dedos.
Las intervenciones comienzan sobre los 6 meses de edad y la reconstrucción total acaba hacia los tres años. El objetivo de la cirugía en los pies es similar que en la situación anterior, pretende conseguir un apoyo adecuado para evitar lesiones y dolores musculares por las malformaciones.
Además de las distintas cirugías, también necesitan controles temporales con:
- – Odontólogo y Fonoaudiólogo o foníatra.
- – Oftalmólogo y Otorrinolaringólogo.
- – Psicomotricista y terapeuta ocupacional, que le ayuden a potenciar el uso funcional de las manos, corregir la postura y la marcha, y a realizar actividades de equilibrio estático, etc.
Existen líneas de investigación sobre opciones de tratamiento médico. Una de estas es la proteína Noggin; dicha proteína antagoniza a la proteína ósea morfogenética tipo 4 (BMP4 por sus siglas en inglés). La proteína Noggin previene el cierre de las suturas craneales y su regulación se encuentra a la baja en el Síndrome de Apert. También se tiene la línea del calphostin C, que es un inhibidor de la proteincinasa C, elemento crucial en la señalización anómala en el Síndrome de Apert. El calphostin actúa mediante la modificación covalente del dominio regulatorio de la unión lipídica de la proteincinasa C, inhibiendo el cierre prematuro en las suturas craneales en modelos animales.
Ámbito psicopedagógico: Los programas de estimulación temprana, entrenamiento en habilidades sociales y de comunicación, seguimiento psicopedagógico y psicoterapia, resultan muy beneficiosos. Además, los procedimientos médicos dolorosos a los que tienen que someterse estos niños pueden acompañarse de intervenciones psicológicas que ya han demostrado su eficacia en el tratamiento del dolor pediátrico. El enfoque psicológico más utilizado para el tratamiento del dolor, el estrés y la ansiedad asociados a procedimientos médicos dolorosos, ha sido el cognitivoconductual. Estas intervenciones psicológicas suelen estar compuestas por modelado filmado, respiración, imaginación, distracción, ensayo conductual, relajación y refuerzo contingente con la utilización de técnicas de afrontamiento.
Pronóstico: Alcanzan hasta los 50 o 60 años de edad. No procrean. La craneosinostosis amenaza producir ceguera, sordera, y retraso mental.
Bibliografía
- Apert E. De l’Acrocephalosyndactylie. Bull Soc Med 1906; 23: 1310.
- Goodman RM, Gorling RJ. Malfor-maciones en el lactante y en el niño síndrome de Apert. Am J Med Genet 1997; 13: 23-34.
- Margolis S, Siegel IM, Choy A, Breinin GM. Oculocutaneous albinism associated with Apert’s syndrome. Am J Ophthalmol 1977; 84 (6): 830-839.
- CN Chirino y RJ Schwartz. Lesiones dermatológicas del síndrome de Apert. A propósito de un caso clínico. Rev. argent. dermatol. v.91 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2010
- Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet. 2012; 90: 175-200.
- Matsumoto K, Urano Y, Kubb Y, Nakanishi H, Arase S. Mutation of the fibroblast growth factor receptor 2 gane in Japanese patients with Apert Syndrome. Plast Reconst Surg 1998; 101: 307-11.
- Filkins K, Russo JF, Boehmer S, Camous M, Przylepa KA. Prenatal ultrasonographic and molecular diagnosis of Apert
- Syndrome. Prenat Diag 1998; 11: 1081-4.
- Wilkie, A. O. M.; Slaney, S. F.; Oldridge, M.; Poole, M. D.; Ashworth, G. J.; Hockley, A. D.; Hayward, R. D.; David, D. J.; Pulleyn, L. J.; Rutland, P.; Malcolm, S.; Winter, R. M.; Reardon, W. : Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature Genet. 9: 165-172, 1995.
- Solomon LM, Cohen MM Jr, Pruzansky S. Pilosebaceus abnormalities in Apert-type acrocephalosyndactyly. Birth Defects Orig Artic Series 1971; 7: 193- 195.
- Cohen JR, Kreiborg S. Cutaneo manifestations of Apert Sydrome. Am J Med Genet 1999; 58 (1): 94- 96.
Jous U. Funtional treatment of cranio-synostosis during chilhood. Br J Oral Maxillofac Surg 1998; 36: 91-98.
Sarimski K. Children with Apert syn-drome: behavio oral Problems and family stress. Kindercentrum Munchen, Germany. Rev med child neurol, 1998; 40: 44- 49.