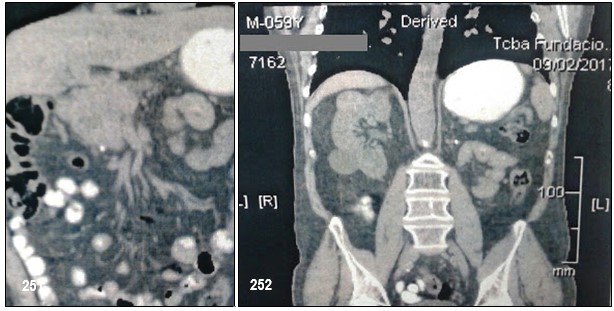
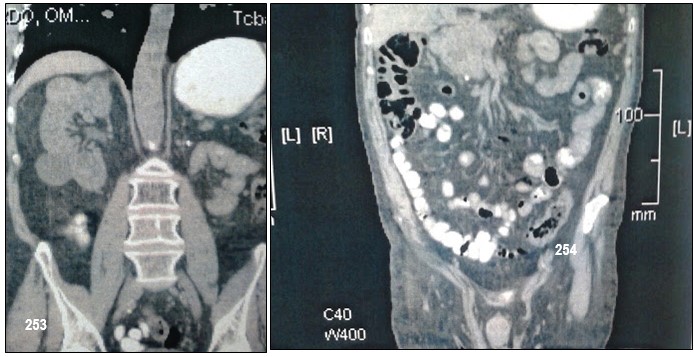
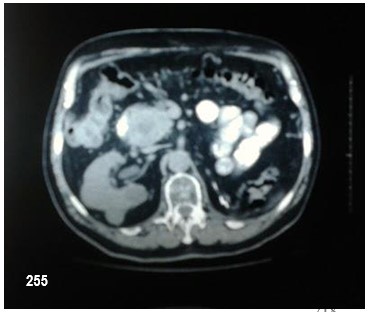
Podríamos resumir diciendo que el paciente, motivo de la presentación, presentó feocromocitoma bilateral, tumor neuroendócrino pancreático y carcinoma renal de células claras. El análisis genético fue positivo para VHL. Entre los antecedentes familiares, madre fallecida por probable hemagioblastoma, un hijo con feocromocitoma bilateral fallecido por hemangiomatosis cerebelosa. Enfermedad de Von Hipell Lindau.
Los Editores del Hipertexto agradecen la gentileza del Dr. Juan Pedro Macaluso (Argentina) por autorizarnos la publicación del caso.
INFORMACIÓN BÁSICA: Enfermedad de Von Hippel-Lindau (EVHL), (ORPHA:892/CIE-10:Q85.8/OMIM:193300).- La VHL es un síndrome familiar de predisposición al cáncer, asociado a una variedad de tumores benignos y malignos, principalmente tumores de retina y de cerebelo, y al hemangioblastoma espinal, carcinoma de células renales (CCR) y feocromocitoma.
La EVHL es una alteración multisistémica caracterizada por la aparición de neoplasias múltiples, entre las que se incluyen tumores de crecimiento anormal de los vasos retinianos como son hemangioblastomas de la retina y del sistema nervioso central (SNC) (principalmente del cerebelo y la médula espinal), carcinoma renal de células claras, feocromocitoma, quistes del páncreas, riñón, hígado y epidídimo y, según se recoge en un estudio muy reciente, tumores del saco endolinfático y del ligamento redondo. Considerada una enfermedad rara de carácter hereditario autosómico dominante, dentro del grupo de las facomatosis. La manifestación inicial más frecuente es la angiomatosis en la retina y los hemangioblastomas en el cerebelo, apareciendo posteriormente tumores en cerebro, médula espinal, feocromocitoma, cistoadenoma seroso microquístico de páncreas y carcinomas de células renales.
Se clasifica en dos tipos en función de la presencia o ausencia de feocromocitoma. El diagnóstico de sospecha se basa en la clínica y los antecedentes familiares y se confirma mediante estudio molecular. Los criterios diagnósticos incluyen:
- a) más de un hemangioblastoma en el SNC,
- b) un hemangioblastoma y manifestaciones viscerales de la enfermedad, y
- c) cualquier manifestación en el SNC o visceral y antecedentes familiares positivos.
Actualmente se considera que el tratamiento más eficaz es la prevención de las complicaciones relacionadas con crecimientos tumorales. Este enfoque precisa de un diagnóstico presintomático y un seguimiento periódico a lo largo de la vida, realizado por un equipo multidisciplinario, en el que las diferentes técnicas de imagen juegan un papel muy importante
Los primeros casos descritos de este proceso se remontan a hace más de un siglo, aunque se conoce con este nombre sólo desde 1932. Eugene Von Hippel fue el primero en describir, en 1904, como hemangioblastomas los tumores de la retina. Arvid Lindau, en 1926, fue quien de modo definitivo asoció los hemangiomas retinianos con los del SNC y los procesos viscerales.
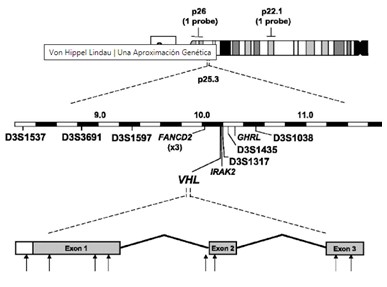
Se trata de una enfermedad con una herencia autosómica dominante, de tumores multisistémicos, de alta penetrancia, determinada por la alteración de un gen (gen VHL) localizado en el brazo corto del cromosoma 3 (3p25.5). Las mutaciones heterogéneas de la línea germinal del gen VHL (3p25-26), que generalmente surgen de novo, se encuentran en 70 a 80% de los individuos. Este gen tiene las características de un gen supresor tumoral, que responde al modelo two-hits. Según este modelo, en los procesos cancerosos hereditarios debidos a un gen supresor tumoral, los pacientes heredan del padre afectado el alelo mutado (mutación germinal) (first-hit), que les predispone a desarrollar el tumor, pero se precisa una segunda mutación en el alelo normal, que procede del padre sano (mutación somática) (second-hit), para que el tumor aparezca, ya que sólo cuando las 2 copias de un gen supresor tumoral están inactivadas, aparece la neoplasia. La alteración del alelo normal se produciría por mutaciones puntuales o por hipermetilación.
El gen VHL se localizó en el brazo corto del cromosoma 3 en 1988, fue clonado en 1993 y está compuesto por tres exones. Estos tres exones codifican una proteína VHL, de aproximadamente 30 kD, compuesta por 213 aminoácidos. Esta proteína se une a las proteínas denominadas elonguinas B y C y cullina 2, que regulan la elongación de la transcripción mediada por la ARN-polimerasa II. Las mutaciones en el gen VHL comprometen la capacidad de unión de la proteína VHL a las elonguinas B y C, siendo ésta incapaz de secuestrarlas, lo que estimula la actividad de la ARN-polimerasa II y aumenta la transcripción génica. De todos modos, todavía no se conocen cuáles son los genes regulados por la proteína VHL, que probablemente se activan sólo en algunos tejidos específicos, en los que se desarrollan los tumores. Las elonguinas y la cullina parecen actuar, especialmente, sobre proteínas reguladoras de algunos ARN mensajeros (ARNm) inducibles por hipoxia (como los ARNm del factor de crecimiento endotelial o del factor de permeabilidad vascular), lo que se ha relacionado con la naturaleza altamente vascularizada de los tumores de EVHL y con su capacidad para producir péptidos angiogénicos.
Epidemiología.- La EVHL es un proceso infrecuente con una incidencia estimada de entre 1/36.000 y 1/45.000 nacidos vivos. La prevalencia de portadores se ha calculado entre 1/53.000 y 1/85.000 habitantes, siendo más elevada (1/39.000) en la Selva Negra alemana, donde las distintas familias afectadas parecen tener un ancestro común.
Manifestaciones clínicas.- La expresividad clínica de la EVHL es muy heterogénea. Cada familia afectada puede presentar diferentes manifestaciones de la enfermedad, según los distintos tipos y localizaciones de las mutaciones del gen.
National Cancer Institute de EE.UU. propuso en 1995 la clasificación de la EVHL en dos tipos:
- VHL tipo 1, cuando no presenta feocromocitoma, y tipo 2, cuando lo presenta.
- VHL tipo 2: alto riesgo de feocromocitoma (exclusivamente mutaciones missense)
- Tipo 2A: bajo riesgo de carcinoma de células renales
- Tipo 2B: alto riesgo de carcinoma renal
- • Tipo 2C: únicamente feocromocitoma
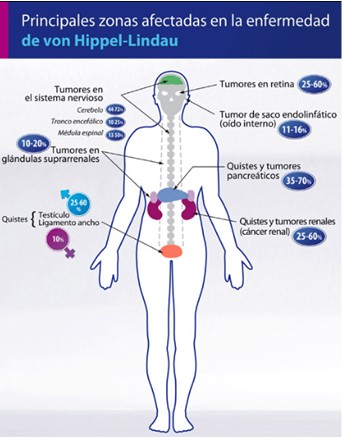
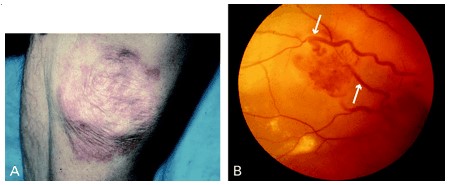
La edad de aparición de las lesiones tumorales (hemangiomas, feocromocitomas, carcinoma renal, etc.) es mucho más temprana que cuando estos procesos se presentan aislados. El 80% de las lesiones son clínicamente aparentes antes de los 40 años y a los 50 años el 95% de los afectados ya ha desarrollado una o más alteraciones. A pesar de tratarse de un síndrome pluritumoral, en algunas series, la mitad de los afectados presenta alteración en un solo órgano.
Según la revisión de Neumann y Wiestler, en la que se recogen las características de las familias con más de 8 miembros afectados, las manifestaciones más frecuentes son los hemangioblastomas del SNC y los angiomas de la retina que aparecen entre el 8 y el 100% de los casos. Los hemangioblastomas del SNC son tumores generalmente quísticos, de crecimiento lento. Aparecen en el cerebelo (el 60-80% de casos), el bulbo raquídeo o la médula espinal, raramente son supratentoriales o se localizan en el nervio óptico y, en ocasiones, pueden ser múltiples. Clínicamente, pueden presentarse como un síndrome cerebeloso, con hidrocefalia, hipertensión arterial y endocraneana, sangrado o siringomielia. Según un registro regional de tumores en el norte de Inglaterra, el 14% de los hemangioblastomas del SNC forman parte de una EVHL y constituyen, junto con el carcinoma renal, las causas de muerte más frecuentes de los pacientes con EVHL.
Los hemangiomas retinianos son una de las primeras alteraciones en aparecer, incluso durante la primera década de la vida, aunque sus manifestaciones clínicas son posteriores. Son tumores vasculares benignos, múltiples en más de la mitad de los casos, bilaterales (el 20% de los casos) y recurrentes. Si no se identifican en fases tempranas y se tratan, evolucionan a la ceguera del ojo afectado, por hemovítreo.
El carcinoma renal de células claras se presenta aproximadamente en el 40% de los casos (entre el 13 y 91%). A diferencia de los casos aislados, no es más frecuente en los varones y puede ser multifocal. En las series más antiguas, se diagnosticaban con metástasis en el 50% de los casos y eran la causa de muerte en un tercio de los individuos afectados por la EVHL2. Los quistes renales, también muy frecuentes, a menudo albergan carcinomas.
Los feocromocitomas se presentan aproximadamente en el 20% de los pacientes20, aunque existen familias de EVHL en las que el feocromocitoma es mucho más frecuente, e incluso puede ser la única manifestación de la enfermedad. En este último caso, se ha sugerido clasificar a los pacientes como tipo 2C. Pueden ser adrenales o extraadrenales y son generalmente benignos, aunque existen casos de malignidad.
La presentación clínica del feocromocitoma en la EVHL se diferencia de los casos aislados en que son de aparición temprana, incluso aparecen durante la primera década4, bilaterales (50%) y, en ocasiones, poco sintomáticos. Si constituyen la primera manifestación de la EVHL, pueden confundirse con casos aislados o con feocromocitomas familiares.
Los quistes pancreáticos y hepáticos parecen menos frecuentes, quizá porque no todas las series los registran (entre el 7-60% de los casos) y son de comportamiento generalmente benigno. En el páncreas, los cistadenomas pueden ser múltiples, afectando a toda la glándula.
El cistadenoma del epidídimo o delligamento ancho aparece en la mitad de los varones afectados y se localiza principalmente en la cabeza del conducto. Pueden ser palpables, de tamaño en general inferior a los 2 cm, sólidos o quísticos, uni o bilaterales y de curso benigno.
En las mujeres, se han descrito algunos casos de cistadenomas papilares en el ligamento ancho, de características histológicas muy semejantes a las descritas en el epidídimo.
La última lesión que se ha asociado a la EVHL ha sido el tumor del saco endolinfático. Cuando se estudió detenidamente la función auditiva de una serie de 121 pacientes con EVHL, la prevalencia de trastornos auditivos fue muy elevada (> 65% de casos y bilateral en > 50%), con una incidencia de tumores del saco endolinfático del 11%,
La bibliografía recoge otras lesiones esporádicas en el tiroides, el pulmón, el epiplón, meninges, el hueso, la piel, etc., que no se han llegado a establecer como manifestaciones propias de la enfermedad.
Los tumores renales y los hemangioblastomas pueden acompañarse de un aumento de eritropoyetina, asociado a la policitemia, que presentan entre el 10-20% de los hemangioblastomas y a la producción de factores de crecimiento y permeabilidad vascular, a los que se atribuye la hipervascularización de dichos tumores.
Diagnóstico y seguimiento clínico.- Hasta el descubrimiento del gen causante de la enfermedad, el diagnóstico se realizaba, siguiendo los criterios establecidos por Melmon y Rose en 1964, en aquellos pacientes con más de un hemangioblastoma del SNC o con un único hemangioma, asociado a otra de las manifestaciones viscerales de la enfermedad, y en aquellos que presentaban una sola manifestación, cuando existía historia familiar positiva. Neumann et al, en 1993, diagnosticaron clínicamente la EVHL en el 19% de los pacientes afectados de feocromocitomas, por lo que recomendaron descartarla en todos los feocromocitomas aislados. Posteriormente, al analizar las mutaciones en dicho grupo de pacientes, se descubrió que la gran mayoría tenían la misma mutación y que existía entre ellos un efecto fundador que explicaba el alto porcentaje de afectados.
Recientemente, Brauch y Bar, en sujetos con feocromocitoma esporádico, sin antecedentes familiares, sólo encontraron mutaciones del gen VHL en un 3,7 y un 3% de los casos, por lo que sostienen que la EVHL es rara y no debe descartarse clínicamente en los casos de feocromocitoma aislado.
Cuando se siguen pacientes con EVHL o se estudia a los portadores de la mutación, la historia de los antecedentes familiares debe ser detallada, ya que el tipo de manifestación clínica y la cronología de su presentación dependen de cada familia. Actualmente, muchas de las manifestaciones pueden y deben diagnosticarse antes de su expresividad clínica. Los hemangioblastomas del SNC se diagnosticarán por resonancia magnética nuclear (RMN) con contraste y estudio angiográfico, el feocromocitoma por determinación periódica de catecolaminas en orina, los hemangiomas retinianos por oftalmoscopia indirecta, el carcinoma renal y los quistes pancreáticos y hepáticos por tomografía computarizada (TC), y el cistadenoma del epidídimo por ecografía. En cuanto al tumor del saco endolinfático, los pacientes al diagnóstico deben ser estudiados detenidamente por ORL, con estudios audiométricos, potenciales evocados auditivos y RMN del peñasco del temporal.
El seguimiento de los individuos clínicamente afectados y de los portadores asintomáticos de la mutación debe abordarse desde una perspectiva multidisciplinaria y mantenerse desde el diagnóstico hasta los 50 años, ya que es raro que aparezcan manifestaciones por encima de esta edad, aunque se han descrito casos de portadores que no manifiestan la enfermedad a los 65 años. La pauta de exploraciones sistemáticas indicada es la siguiente: estudio del fondo del ojo anual por oftalmoscopia indirecta desde los 5 años, determinación anual de catecolaminas en orina desde los 5 años, RMN craneal bianual desde los 15 años, ecografía o TC abdominal bianual a partir de los 20 años, y estudio auditivo, con una periodicidad todavía no establecida, con exploración por RMN del peñasco del temporal en cuanto aparezcan síntomas auditivos; tampoco está establecida la edad para practicar la ecografía para descartar los cistadenomas del epidídimo. La aparición de cualquier síntoma o alteración en las exploraciones sistemáticas llevará a agotar las posibilidades diagnósticas. Las imágenes de lesiones renales, cuando aparecen, deben seguirse mediante TC semestral.
Diagnóstico molecular.- Confirma el diagnóstico clínico y permite identificar a los familiares asintomáticos. Debe realizarse en los casos índices y en sus familiares directos. Las mutaciones en el gen de la EVHL se detectan sólo en el 80% de las familias afectadas. Hasta la fecha, se han descrito más de 500 mutaciones (http://www.umd.necker.fr). Este gran número de mutaciones dificulta la correlación del genotipo con las diferentes manifestaciones fenotípicas. Cada mutación puede llegar a tener una expresión fenotípica característica. Se han descrito muy pocas mutaciones iguales en más de dos familias, lo que indica que tienen un origen reciente, ya que de lo contrario se identificaría la misma mutación en un mayor número de familias, con un probable ancestro común. De acuerdo con esta observación, se han descrito familias con mutaciones aparecidas de novo.
Genética.-
- Herencia autosómica dominante (AD) con 90% penetrancia
- Gen causal: VHL que es un gen supresor tumoral (TSG)
- Gen locus: cromosoma 3 (3p25)
- Producto proteico: pVHL
- Función: ensamblaje fibronectina estabilidad de microtúbulos regulación de HIF (factor inducible por hipoxia)
- En 70- 80% de carcinoma de células claras esporádicos la inactivación somática de VHL conlleva pérdida de expresión pVHL
Funciones de la proteína VHL:
- GLUT 1 (Transportador de glucosa)
- Eritropoyetina
- AC9 (Anhidrasa carbonica 9)
- Controla degradación de la matriz extracelular
- Diferenciación de las células renales
- Maduración y mantenimiento de Túbulos Renales Proximales
El producto de la proteína del gen VHL tiene una longitud de 213 aminoácidos y su funcionamiento se relaciona con la proteína elongina-B y elongina-C. Esta relación en el complejo VHL-elongina B-elongina C mantiene la ligazón entre las proteínas claras especificadas y es la base de su ubicuitinación (el complejo tiene un nivel de estructura proteínica E3). Se afirmó que los sustratos del complejo VBC son las proteínas claras HIF1α y HIF2α, junto a la proteína clara atípica λ (de kinaza??); cuando esta ligadura tiene lugar se establece la proteína clara domena β VHL; por ello la proteínas claras quedan sujeta a ubicuitinación y determinadas por la misma, degradándose a proteasomas.
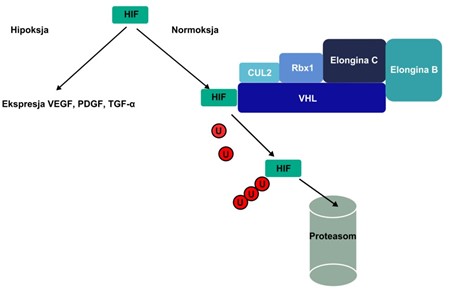
El gen VHL trabaja con otras proteínas para formar el complejo VCB-CUL2. Este complejo causa que otras proteínas en la célula se descompongan si están dañadas o ya no se necesitan. Uno de los enfoques del complejo VCB-CUL2 es ‘hypoxia-inducible factor-2-alpha (HIF-2a). HIF-2a coordina las respuestas del cuerpo a cambios en los niveles de oxígeno mediante el control de la división celular y la formación de vasos sanguíneos y glóbulos rojos nuevos. Cuando los niveles de oxígeno están normales, el complejo VCB-CUL2 detiene a HIF-2a. Cuando VHL se muta, el complejo VCB-CUL2 no puede funcionar propiamente y no puede degradar a HIF-2a o a las otras proteínas cuando estén defectuosas. Entonces, HIF-2a puede estimular excesivamente la división celular y la creación de vasos sanguíneos, cual puede llevar a la formación de tumores y quistes, ambos característicos del síndrome de Von Hippel-Lindau.
UbiquitIn ligasas tipo SCF. El producto génico VHL (von Hippel-Lindau), pVHL, (complejos de ohmios con elongln C, elorigin B y ailin-2, con estructura estructuralmente similar a SCF (proteína Skpl-Cdc53-F-Box), gases de ubiguitina ‘levadura. Estas proteínas diana de potyubiguitylate que luego se degrada por la fteasina. El dominio pVHL a se asemeja a un motivo F-box, que se une a Skpl. El dominio pVHL 13 tiene características de un sitio de acoplamiento de subsnatos. Ub, ubiquitina. Siendo la función más importante de la proteína VHL, inhibir la HIF Factor Inducible por Hipoxia (HIF).
La pVHL interactúa con otras tres proteínas, elongina C y B y Cullina 2 (CUL2), en un complejo denominado VCB-CUL2. pVHL tiene dos dominios estructurales principales: un dominio N-terminal compuesto principalmente de láminas b (el dominio b) y un dominio C-terminal más pequeño entre los aminoácidos 155-192 compuesto principalmente de hélices (dominio a). El dominio a consiste en tres hélices a que se combinan con una cuarta a hélice donada por elongina C. El dominio b está en el lado opuesto del dominio a y es libre de contactar otras proteínas.
Expresión: El pVHL se expresa ampliamente en tejidos humanos fetales y adultos.
Localizacion: El pVHL es en gran medida una proteína citoplasmática, pero parece desplazarse entre el citoplasma y el núcleo. pVHL interactúa con otras tres proteínas, elongina C y B y Cullina 2 (CUL2), en un complejo denominado VCB-CUL2. pVHL tiene dos dominios estructurales principales: un dominio N-terminal compuesto principalmente de láminas b (el dominio b) y un dominio C-terminal más pequeño entre los aminoácidos 155-192 compuesto principalmente de hélices (dominio a). El dominio a consiste en tres hélices a que se combinan con una cuarta a hélice donada por elongina C. El dominio b está en el lado opuesto del dominio a y es libre de contactar otras proteínas.
VHL y angiogénesis: Una función principal del pVHL es regular negativamente los ARNm inducibles por hipoxia, como el ARNm que codifica VEGF, EPO, PDGF y el transportador de glucosa GLUT-1. El pVHL desempeña un papel crítico en el objetivo del factor de transcripción inducible por hipoxia HIF-1a para la degradación por el proteasoma. HIF-1a contribuye a formar el complejo transcripcional HIF-1 responsable de la activación de genes implicados en el metabolismo, la angiogénesis y la apoptosis. El complejo VCB-CUL2 se ha demostrado como un sistema de ubiquitina-ligasa que presenta muchas similitudes con el SCF sistema («proteína Skp1-CUL1-Fbox»). El HIF normalmente se degrada en condiciones normóxicas y la unión a VHL depende de la hidroxilación de Pro 564 en HIF-1a (Figura 1). Cuando el gen VHL está mutado, la ausencia de degradación de HIF es responsable de la acumulación anormal de VEGF y otros ARNm inducibles por hipoxia que explican el fenotipo angiogénico de los tumores VHL. pVHL también puede regular negativamente la producción de VEGF mediante unión directa e inhibición al activador transcripcional SP1.
En ratones homocigotos con VHL inactiva, los embriones morirán temprano debido a un trastorno importante de la vasculogénesis placentaria
Otras funciones, pVHL juega un papel en:
1- la capacidad de las células para salir del ciclo celular y entrar en el estado de reposo.
2- montaje de matriz de fibronectina extracelular.
3- degradación de TGFa LYT10, TGFb y anhidrasas carbónicas CA9 y CA12.
4- regulación del sistema activador de plasminógeno de tipo uroquinasa.
5- inhibición de la invasión inducida por el factor de crecimiento de hepatocitos en el carcinoma de células renales.
6- también se ha demostrado recientemente una interacción directa con la proteína quinasa C atípica C (PKC) z y l.
Por lo tanto, la BVS aparece como un gen multifuncional y puede desempeñar un papel de portero especialmente en el riñón
Homologia: La estructura de secuencia primaria de pVHL muestra una homología mínima con cualquier proteína conocida, pero la conservación evolutiva de pVHL es muy fuerte a excepción de los primeros 53 aminoácidos.
Mutaciones: Las mutaciones de la línea germinal causan la enfermedad de von Hippel-Lindau. Las mutaciones de VHL son heterogéneas.
Germinal: las mutaciones de la línea germinal causan la enfermedad de von Hippel-Lindau. Las mutaciones de VHL son heterogéneas y se distribuyen ampliamente por toda la secuencia de codificación, excepto 5 ‘para el sitio de inicio de traducción para pVHL. Hay algunas mutaciones recurrentes y solo se conoce un efecto fundador, originario de Alemania (T292C que resulta en una sustitución Tyr98His).
Las mutaciones puntuales ocurren en aproximadamente el 60% de los casos (Figura 2) y las deleciones grandes en aproximadamente el 40%. VHL 1 (sin feocromocitoma) se produce principalmente por mutaciones responsables de la proteína truncada (deleciones, mutaciones de desplazamiento de marco y mutaciones sin sentido). La BVS tipo 2 (con alto riesgo de feocromocitoma) se produce principalmente por mutaciones sin sentido. El tipo 2B es la forma potencialmente «completa» de la enfermedad (mutaciones frecuentes: Arg167Gln, Arg167Trp). El tipo 2A está asociado con un riesgo muy bajo de cáncer de células renales de células claras (CCR) (mutación común: Tyr98His). El tipo 2C se caracteriza por la aparición de feocromocitoma solamente (ejemplo: Leu188Val).
Entre el 10 y el 15% de los casos crípticos de VHL podrían explicarse por mutaciones de novo y hay algunos casos mosaicismo de la línea germinal.
Existe alguna evidencia de que los modificadores genéticos pueden influir en la expresión fenotípica de la enfermedad.
A nivel somatico, encuentran mutaciones en el 60% de los CCR esporádicos de células claras. Además, el 15% de los tumores muestran evidencia de inactivación por metilación. Las alteraciones de la BVS se han asociado con la exposición laboral al tricloroetileno.
Somáticas: también son frecuentes en el hemangioblastoma esporádico del SNC, pero son más raras en los tumores esporádicos del saco endolinfático, los cistadenomas serosos pancreáticos y los tumores endocrinos, los cistadenomas epididimarios y los feocromocitomas
La ubiquitina es el elemento central de un conjunto de interacciones entre proteínas que está relacionado con funciones tan importantes como la regulación del ciclo celular, la replicación y transcripción del ADN, la dinámica de la cromatina y otras. La intervención del sistema de la ubiquitina en estos y otros procesos puede ser de dos formas: como modulador de la actividad de proteínas y como marcador de proteínas para su degradación. En este momento solo se hará referencia al segundo caso.
La ubiquitina es una proteína de 78 aminoácidos cuya secuencia se encuentra conservada desde la levadura hasta el hombre. El primer paso del proceso consiste en la unión de la ubiquitina a una enzima activadora de ubiquitina (UBA o E1) mediante un enlace tioester y dependiente de ATP. En el segundo paso la ubiquitina se tranfiere a la enzima conjugante de ubiquitina (E2) también mediante un enlace tioéster. En el tercer paso intervienen las ubiquitinas-ligasas (E3) que, dependiendo del tipo, bien transfieren la ubiquitina desde la E2 hacia un sustrato específico o bien aceptan la ubiquitna desde la E2 y lo transfieren a la proteína sustrato por un residuo de lisina. A esta ubiquitina se une una segunda y a esta una tercera y posteriormente una cuarta hasta formar la poliubiqutina, que es la marca necesaria para que esa proteína sea degradada por un complejo multiproteínico llamado proteasoma de 26 S (para una descripción más completa de las funciones de la ubiquitina y su contribución en la carcinogénesis se invita a los lectores consultar las referencias.
Las mutaciones se han descrito en los tres exones del gen, especialmente en el 1 y el 3: extremo 5′ del exón 3 y 3′ del exón 1, siendo más raras en el exón 2. Las mutaciones son distintas según se trate de familias tipo 1 o tipo 2. Las mutaciones descritas en familias tipo 1 son microdeleciones, inserciones, macrodeleciones, mutaciones que dan lugar a un cambio en el marco de lectura y mutaciones que producen un codón de paro, situadas frecuentemente en el exón 1. En las familias tipo 2, se identifican mutaciones puntuales con mayor frecuencia (entre el 85-96% de los casos), especialmente en el exón 3, en los nucleótidos 712 o 713, que resultan en la sustitución de la arginina por triptófano o glutamina en el codón. Este tipo de mutación se asocia a un riesgo para desarrollar feocromocitoma del 62%. Las mutaciones en los codones situados en los extremos del gen, probablemente, cambian poco la conformación de la proteína VHL y se asocian a las familias tipo 2, con escasas manifestaciones, a excepción del feocromocitoma, que puede ser la única manifestación de la enfermedad.
Distintas mutaciones en el mismo codón provocan cambios que dan lugar a diferentes aminoácidos y a distintas expresiones fenotípicas, lo que puede reflejar un fenotipo específico para cada distinta sustitución de aminoácidos o un fenotipo variable, o simplemente, que el fenotipo no esté totalmente expresado en el momento del estudio genético.
El gen VHL mutado se ha descrito no sólo en los pacientes afectados de EVHL, sino también en el tejido tumoral de los carcinomas de células claras, en hemangioblastomas del SNC y en cistadenomas del epidídimo de casos esporádicos.
Aunque la incidencia de la EVHL en los casos de feocromocitoma aislado sea baja y se haya aconsejado limitar el análisis genético a los casos de presentación clínica atípica (familiar, temprana y múltiple), como indican Brauch et al, valorando el coste-beneficio del diagnóstico de un caso índice, puede estar indicado realizar dicho estudio molecular en los casos de feocromocitoma esporádico y probablemente la misma consideración deba aplicarse a otros de los tumores relacionados con la EVHL (hemangioblastomas, angiomas retinianos o carcinoma renal). El diagnóstico molecular permite, además, el consejo genético a los afectados por la mutación.
Como inconveniente del diagnóstico molecular, cabe señalar que entre el 20-38% de las mutaciones escapan a la detección, especialmente en familias con EVHL tipo 1.
Métodos diagnósticos.- El diagnóstico puede establecerse por la presencia de un único tumor típico (p. ej. hemangioblastoma retiniano o del SNC, o CCR) y una historia familiar positiva de VHL. Si no hay historia familiar (sobre un 20% de novo), son necesarios tumores múltiples para el diagnóstico. El hemograma completo, la determinación en orina de metabolitos de catecolaminas, el análisis de orina, y la citología de orina pueden ser indicativos de policitemia, feocromocitoma, anomalías renales, y CCR. Los estudios de imagen pueden ser utilizados para detectar tumores del SNC, feocromocitoma, tumores del saco endolinfático, tumores renales y quistes pancreáticos.
Diagnóstico diferencial.- El diagnóstico diferencial incluye: neoplasia endocrina múltiple, neurofibromatosis, enfermedad poliquística renal, esclerosis tuberosa, síndrome de Birt-Hogg-Dube, y síndromes feocromocitoma-paraganglioma hereditarios (consulte estos términos), asociados a mutaciones de la subunidad succinato deshidrogenasa (SDHB, SDHC and SDHD).
Diagnóstico prenatal.- El diagnóstico prenatal es posible si se ha identificado la mutación responsable en un miembro afecto de la familia.
Consejo genético.- El patrón de herencia es autosómico dominante. Debe ofrecerse consejo genético.
Tratamiento.- Por tratarse de procesos neoplásicos, el tratamiento debe ser quirúrgico y supone la implicación de distintas especialidades para cada proceso. Cuanto más temprano sea el diagnóstico, menor será el tamaño del tumor, la resección quirúrgica podrá ser más radical y supondrá menor riesgo de complicaciones. Los hemangioblastomas del SNC se intervendrán, en función de su tamaño y localización, cuando las lesiones sean sintomáticas. La radioterapia o la radiocirugía pueden ser útiles en los casos no extirpables o residuales. Los hemangioblastomas de la retina son susceptibles de fotocoagulación y crioterapia, especialmente efectivas en lesiones pequeñas o de vitrectomía si existe sangrado.
El carcinoma renal de células claras precisa de una cirugía conservadora con escisiones lo menores posible, ante la posible aparición de nuevos focos tumorales. La cirugía radical debe reservarse para tumores de más de 5 cm. En los casos de nefrectomía bilateral que se someten a trasplante, no se ha descrito que la terapia inmunosupresora aumente el riesgo de recurrencia tumoral. El feocromocitoma tiene el mismo tratamiento que en los casos esporádicos, siendo preciso descartarlo en todos los pacientes afectados de la EVHL, antes de la intervención por cualquier otro proceso.
Los tumores del saco endolinfático deben resecarse completamente, de lo contrario existe un alto índice de recurrencias, aunque el riesgo de pérdida auditiva por iatrogenia quirúrgica es considerable, si el tumor no es muy pequeño. Los quistes hepáticos, pancreáticos y del epidídimo son raramente sintomáticos y no suelen intervenirse.
Pese a los grandes progresos que se han producido en los últimos 5 años en el conocimiento de esta enfermedad, es preciso desentrañar los mecanismos de actuación de la proteína del gen VHL para desarrollar nuevas estrategias terapéuticas dirigidas a inhibir el crecimiento tumoral. Por el momento, el diagnóstico temprano de los casos índice y el diagnóstico molecular de portadores y su seguimiento estricto son los mejores medios para combatirla. Puesto que las lesiones tumorales de la EVHL pueden ser aisladas, asincrónicas y de diagnóstico y de estudio por especialidades diversas, la enfermedad puede pasar desapercibida en los casos índices. Hay que considerar el diagnóstico de EVHL en todos los pacientes con hemangioblastomas del SNC o la retina, feocromocitomas familiares o bilaterales, carcinomas renales familiares o multicéntricos o de aparición temprana y tumores del saco endolinfático bilaterales.
Bibliografía
- Manski TJ, Heffner DK, Glenn GM,Patronas NJ,Pikus AT,Katz D et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA, 277 (1997), pp. 1461-1466 Medline
- Huson SM,Harper PS,Hourihan MD,Cole G,Weeks RD,Compston DAS. Cerebellar haemangioblastoma and Von Hippel-Lindau disease. Brain, 109 (1986), pp. 1297-1310 Medline
- Melmon KL,Rosen SW. Lindau’s disease: review of the literature and study of a large kindred. Am J Med, 36 (1964), pp. 595-617 Medline
- Pheochromocytoma in Von Hippel-Lindau disease: clínicasl presentation and mutation analysis in a large, multigenerational kindred. J Clin Endocrinol Metab 1998; 117-120
- Knudson AG Jr. Antioncogenes and human cancer. Proc Natl Acad Sci USA, 90 (1993), pp. 10914-10921 Medline
- Prowse A,Webster A,Richards F,Richard S,Olschwang S,Resche F et al. Somatic inactivation of the VHL gene in Von Hippel-Lindau disease tumores. Am J Genet, 60 (1997), pp. 765-771
- Seizinger BR,Rouleau GA,Ozelius LJ,Lane AH,Farmer GE,Lamiell JM et al. Von Hippel-Lindau disease maps to the region of chromosoma 3 associated with renal carcinoma. Nature (Lond), 332 (1988), pp. 268-269
- Kaelin WG Jr. «Molecular basis of the VHL hereditary cancer syndrome.» Nat Rev Cancer (2002). 2(9):673-82.
- Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep. 2009 Mar;11(2):94-101.
- Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruine AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010 Jun;221(2):125-38. doi: 10.1002/path.2689.
- VHL. Genetics Home Reference. National Institutes of Health. U.S. Department of Health and Human Services. Reviewed August 2012, Published August 2015
- Shahzad H, Kehar SI, Ali S, Tariq N. Expression of Von Hippel – Lindau (VHL) gene mutation in diagnosed cases of renal cell carcinoma. Pak J Med Sci. 2014 Jul;30(4):880-5.
- Takagi Y,Pause A,Conaway RC,Conaway JW. Identification of elongin C sequences required for interaction with the von Hippel-Lindau tumor suppressor protein. J Biol Chem, 272 (1997), pp. 27444-27449 Medline
- Kibel A,Ilopoulos O,De Caprio JA,Kaelin WG. Binding of the Von Hippel-Lindau tumor supressor protein to elongin B and C. Science, 269 (1995), pp. 1444-1446 Medline
- Bertherat J. Von Hippel-Lindau tumor supressor protein and transcription elongation: new insights into regulation of gene expression. Eur J Endocrinol, 134 (1996), pp. 157-159 Medline
- Maher ER,Kaelin WG. Von Hippel-Lindau disease. Medicine (Baltimore), 76 (1997), pp. 381-391
- Iliopoulos O,Levy AP,Jiang C,Kaelin WG,Goldberg MA. Negative regulation fo hipoxia-inducible genes by the Von Hippel-Lindau protein. Proc Natl Acad Sci USA, 93 (1996), pp. 10595-10599 Medline
- Maher ER,Iselius L,Yales JR,Littler M,Benjamin C,Harris R et al. Von Hipple-Lindau disease: a genetic study. J Med Genet, 28 (1991), pp. 443-447 Medline
- Maddock IR, Moran A,Maher ER,Teare H,Norman A,Payne S et al. A genetic register for von Hippel-Lindau disease. J Med Genet, 33 (1996), pp. 120-127 Medline
- Neumann HP.H, Wiestler OD. Clustering of features of Von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet, 337 (1991), pp. 1052-1054
- Newman HP.H, Eng C, Mulligan LM,Glavac D,Zäuner I,Ponder BA et al. Consequences of direct genetic testing for germline mutations in the clínicasl management of families with multiple endocrine neoplasia, type II. JAMA, 274 (1995), pp. 1149-1151
- Brauch H, Hoeppner W, Jähnig HH,Wöhl T,Engelhardt D,Spelsberg F et al. Sporadic pheocromocytomas are rarely associated with germline mutations in the vhl tumor supressor gene or the ret protooncogene. J Clin Endocrinol Metab, 82 (1997), pp. 4101-4104
- Linehan WM, Lerman MI, Zbar B. Identification of the Von Hippel-Lindau gene. JAMA, 273 (1995), pp. 564-570
- Chen F, Kishida T, Yao M, Hustad T,Glavac D,Dean M et al. Germ-line mutations in the Von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat, 5 (1995), pp. 66-75
- Neumann HPH. Basic criteria for clínicasl diagnosis and genetic counselling in Von Hippel-Lindau syndrome. J Vasc Dis, 16 (1987), pp. 220-226
- Chamorro A, Saldaña C, Rodríguez R, García D. Hemangiomas supratentoriales en la enfermedad de Von Hippel-Lindau. Med Clin (Barc), 91 (1988), pp. 759
- Maher ER, Yates JR.W, Harries R, Benjamin C, Harris R,Moore AT et al. Clínicasl features and natural history of Von Hippel-Lindau disease. Q J Med, 77 (1990), pp. 1151-1163
- Annesley WH, Leonard BC, Shields JA, Tasman WS. Fifteen years review of treated cases of retinal angiomatosis. Transact Am Acad Ophthalmol Otolaryngol, 83 (1977), pp. 446-453
- Poston CD, Jaffe GS, Solomon D,Zbar B,Linehan WM,Walther MM. Characterization of the renal pathology of a familial form of renal cell carcinoma associated with von Hippel-Lindau disease: clínicasl and molecular genetic implications. J Urol, 153 (1995), pp. 22-26
- Gross DJ, Avishai N, Meiner V, Filon D,Zbar B,Abeliovich D. Familial pheocromocytoma associated with a novel mutation in the Von Hippel-Lindau gene. J Clin Endocrinol Metab, 81 (1996), pp. 147-149
- Atuk NO, McDonald T, Wood T,Carpenter JT,Walzack MP,Donaldson M et al. Familial pheocromocytoma, hypercalcemia and Von Hippel-Lindau disease: a ten years study of a large family. Medicine (Baltimore), 58 (1979), pp. 209-218
- Ritter MM, Frilling A,Crossey PA,Höppner W,Maher ER,Mulligan L et al. Isolated familial pheochromocytoma as a variant of Von Hippel-Lindau disease. J Clin Endocrinol Metab, 81 (1996), pp. 1035-1037
- Zbar B, Kishida T,Chen F,Schmidt L,Maher ER,Richards FM et al. Germline mutations in the Von Hippel-Lindau disease gene in families from North America, Europe and Japan. Hum Mutat, 8 (1996), pp. 348-357
- Bender BU, Altehofer C,Januszewicz A,Gartner R,Schmidt H,Hoffmann MM et al. Functioning thoracic paraganglioma: association with Von Hippel-Lindau syndrome. J Clin Endocrinol Metab, 82 (1997), pp. 3356-3360
- Burgos R, R, Hernández-Pascual C,Mesa J. Feocromocitoma y enfermedad de Von Hippel-Lindau. Med Clin (Barc), 102 (1994), pp. 797
- Neumann HP.H, Berger DP, Sigmund G, Blum U,Schmidt D,Parmer RJ et al. Pheochromocytomas, multiple endocrine neoplasia type 2 and Von Hippel-Lindau disease. N Engl J Med, 329 (1993), pp. 1531-1538
- Richard S, Beigelman C,Duclos JM,Fendler JP,Plauchu H,Plouin PF et al. Pheocromocytoma as the first manifestation of Von Hippel-Lindau disease. Surgery, 116 (1994), pp. 1076-1081 Medline
- García A, Matías-Guiu X, Cabezas R,Chico A,Prat J,Baiget M et al. Molecular diagnosis of Von Hippel-Lindau disease in kindred with predominance of pheocromocytoma. Clin Endocrinol (Oxf), 46 (1997), pp. 359-363
- Crossey PA, Richards FM,Foster K,Green JS,Prowse A,Latif F et al. Identification of intragenic mutations in the Von Hippel-Lindau disease tumor suppressor gene and correlations with disease phenotype. Hum Mol Genet, 3 (1994), pp. 1303-1308 Medline
- Girelli R, Bassi C, Falcono M, De Santis L,Bonora A,Caldiron E et al. Pancreatic cystic manifestations in Von Hippel-Lindau disease. Int J Pancreatol, 22 (1977), pp. 101-109
- Choyke PL, Glenn GM,Lubensky IA,Lubensky IA,Thakore K,Zbar B et al. Epididymal cystadenomas in Von Hippel-Lindau disease. Urology, 49 (1997), pp. 926-931 Medline
- Werness BA, Guccion JG. Tumor of broad ligament in Von Hippel-Lindau disease of probable mullerian origin. Int J Gynecol Pathol, 16 (1997), pp. 282-285
- Should endolymphatic sac tumors be considered part of the Von Hippel-Lindau complex? Neurosurgery1997;40:848-55
- Brauch H, Kishida T, Glavac D, Chen F, Pausch F,Hofler H et al. Von Hippel-Lindau disease with feocromocitoma in the Black Forest region in Germany: evidence for a founder effect. Hum Genet, 95 (1995), pp. 551-556 Medline
- Bar M, Friedman E,Jakobovitz O,Leibowitz G,Lerer I,Albeliovich D et al. Sporadic phaechromocytomas are rarely associated with germline mutations in the Von Hippel-Lindau and RET genes. Clin Endocrinol (Oxf), 47 (1997), pp. 707-712
- Spetzger U, Bertalanffy H, Huffmann B, Mayfrank L, Reul J,Gilsbach JM. Hemangioblastomas of the spinal cord and the brainsted: diagnostic and therapeutic features. Neurosurg Rev, 19 (1996), pp. 147-151 Medline
- Davies DR, Norman AM, Whitehouse RW, Evans DG. Non-expression of Von Hippel-Lindau phenotype in a obligate gene carrier. Clin Genet, 45 (1994), pp. 104-106
- Escudero D, Moral A, Pou A. El seguimiento en la enfermedad de Von Hippel-Lindau. Med Clin (Barc),93(1989),pp. 319
- Choyke P, Glenn G, Walther M, Zbar B, Weiss G, Alexander RB et al. The natural history of a renal lesions in von Hippel-Lindau disease: a serial CT study in 28 patients. Am J Roentgenol, 159 (1992), pp. 1229-1234
- Beroud C, Joly D, Gallou C, Staroz F, Orfanelly MT, Junien C. Software and database for the analysis of mutations in the Von Hippel-Lindau gene. Nucleic Acids Res, 26 (1998), pp. 256-258 Medline
- Richards FM, Payne SJ, Zbar B, Affara NA, Fergusson-Smith MA, Maher ER. Molecular analysis of the novo germline mutations in the Von Hippel-Lindau disease gene. Hum Mol Genet, 4 (1995), pp. 2139-2143 Medline
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=892