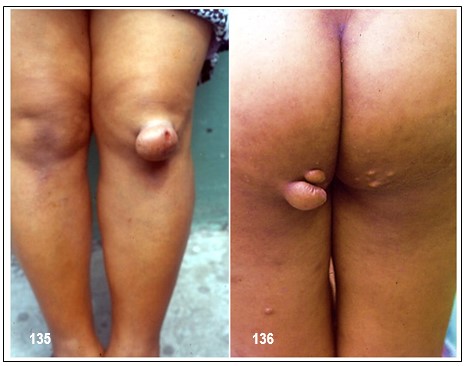
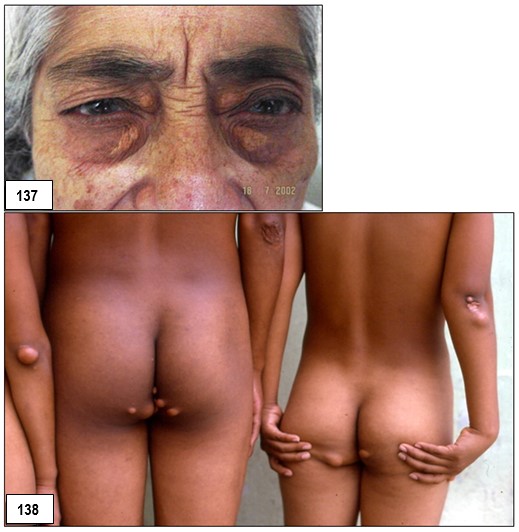
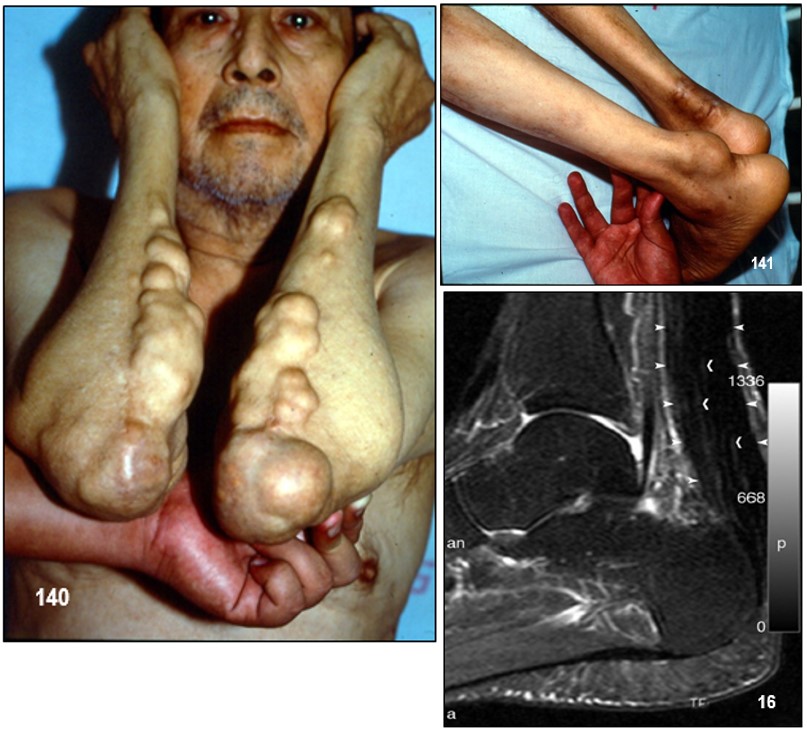
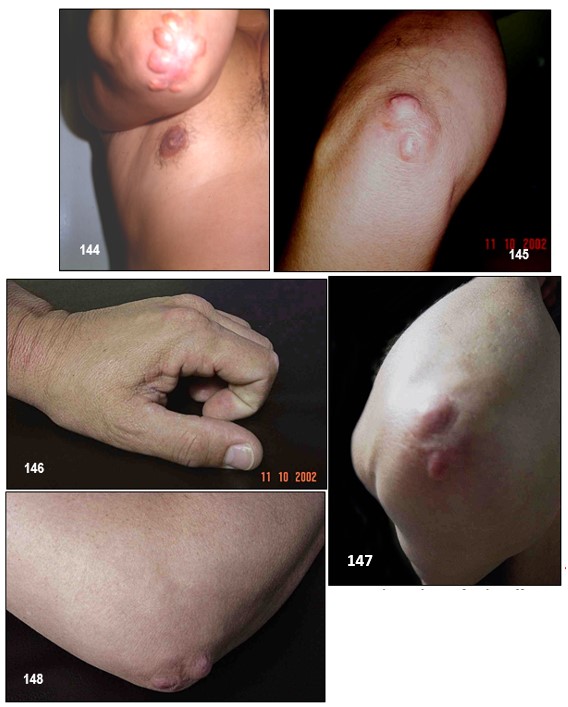
Imagen 130.- Dislipidemias. Hipercolesterolemia familiar homocigota. Caso emblemático.- Primer paciente con hipercolesterolemia familiar homocigota diagnosticado por el autor en 1985. El estudio de la paciente así como de sus progenitores reveló importantes niveles de colesterol total a expensas del transportado por las lipoproteínas de baja densidad (cLDL). Note xantomas tuberosos en los codos y glúteos, y tendinosos en las manos. La imagen no muestra los xantomas tendinosos en ambos tendones de Aquiles. Fuente: Tama Viteri FA. Clínica y Terapéutica de las Dislipidemias. Primera Edición, 2011. UG-Ecuador.Imagen 132.- Hipercolesterolemia familiar heterocigota. Múltiples nódulos de gran tamaño de color amarillo-rojizo en el codo, muy sugestivos de xantomas tuberosos. Imagen 133.- Hipercolesterolemia familiar heterocigota. Varias tumoraciones de aspecto nodular y papular de tamaños diversos que comprometen ambas rodillas y que corresponden a xantomas tuberosos. Imagen 134.- Hipercolesterolemia familiar heterocigota. Xantomas tuberosos. Tumoraciones nodulares en ambos codos.Imagen 135.- Hipercolesterolemia familiar heterocigota.- Nódulo pediculado único de gran tamaño ubicado en la rodilla sugestivo de xantoma tuberoso. Imagen 136.- Hipercolesterolemia familiar heterocigota.- Varios nódulos de aspecto xantomatoso de varios tamaños ubicados en glúteos; corresponde a la paciente de la imagen.Imágenes con fines didácticos: Imagen 137.- Hipercolesterolemia familiar heterocigota. Xantelasmas. Presencia de xantelasmas en el borde libres de los párpados. Imagen 138.- Hipercolesterolemia familiar (fenotipo IIa). Tres hermanos con la forma familiar heterocigota. Obsérvese xantomas tuberosos en codos y región glútea y subglútea. Todos ellos procedentes de la Provincia Peninsular de Santa Elena; es frecuente este tipo de patología en esta zona, probablemente por la alta incidencia de endogamia existente entre sus habitantes. Fuente: Tama Viteri FA. Clínica y Terapéutica de las Dislipidemias. Primera Edición, 2011. UG-Ecuador.Imagen 139.- Hipercolesterolemia familiar (fenotipo IIa). A mayor detalle xantomas tuberosos en codos en dos pacientes de la imagen. Fuente: Tama Viteri FA. Clínica y Terapéutica de las Dislipidemias. Primera Edición, 2011. UG-Ecuador.Hipercolesterolemia familiar heterocigota (fenotipo IIa).- Paciente de 67 años de edad que ingresa por presentar masas tumorales espectaculares ubicadas en miembros superiores, glúteos, rodillas y en el tendón de Aquiles (imagen 2). Refería que a la edad de 5 años aparecieron las primeras tumoraciones en brazos (codos), rodillas y glúteos y que a la edad de 16 años nota éstas en ambos tendones aquilianos. Decía ser el segundo de cuatro hermanos y que sus padres tenían “problemas con el colesterol”; recientemente se había enterado que una hermana había fallecido de infarto agudo al miocardio. Refería dolor anginoso recurrente. A la exploración semiológica destacaban tumoraciones de varios tamaños que formaban una “montaña” en el borde cubital de ambos antebrazos, igualmente en codos muy sugestivas de xantomas tuberosos; estas tumoraciones no tenían aspecto inflamatorio, ni supurativas, ni dolorosas. Con sospecha de hipercolesterolemia primaria se procedió a solicitar exámenes. Colesterol total: 498 mg/dl, triglicéridos: 133 mg/dl, cLDL: 375 mg/dl, cHDL: 33 mg/dl. Aspecto del plasma: claro. ECG: cardiopatía isquémica. Con este perfil lipoproteíco se catalogó la dislipidemia como primaria, compatible con hipercolesterolemia familiar heterocigota; lamentablemente no se pudo contar con ningún familiar para filiar con certeza este trastorno. Imagen 141.- Dislipidemias. Hipercolesterolemia familiar heterocigota (fenotipo IIa). Tumoración de aspecto nodular muy sugestiva de xantoma del tendón de Aquiles. Adviértase que la de lado derecho fue retirada quirúrgicamente con fines estéticos. Imagen 16.- Con fines Didácticos: Imagen de RM del tendón de Aquiles en un paciente con un xantoma. Plano sagital anterior que muestra un engrosamiento difuso y un patrón reticular de los tendones (puntas de flecha abiertas). Las puntas de flecha marcan delinear el tendón de Aquiles. Los tendones de Aquiles normales no superan los 7mm de diámetro en la región entre el calcáneo y el punto donde las fibras del tendón comienzan a irradiar hacia sus orígenes.Imagen 142.- Dislipidemia. Hipercolesterolemia familiar (fenotipo IIa). Paciente de 52 años de edad con antecedentes de hipercolesterolemia familiar heterocigota que ingresa por presentar una placa de xantelasma en el párpado superior derecho, múltiples pápulas de varios tamaños en ambos codos y otras de aspecto tumoral en los glúteos, correspondientes a xantomas tuberosos. Se realizaron perfil lipoproteico: cLDL: 367 mg/dl. ECG: cardiopatía coronaria. Biopsia de xantoma. Imagen143.- Dislipidemia. Hipercolesterolemia familiar (fenotipo IIa). Xantomas tuberosos en ambos codos. Fuente: Tama Viteri FA. Clínica y Terapéutica de las Dislipidemias. Primera Edición, 2011. UG.Imagen 144-145.-Hipercolesterolemia familiar (fenotipo IIa).- Médico de 51 años de edad, con antecedentes de xantomatosis tuberosa y tendinosa por HF heterocigota, que es sometido a terapia intensa con estatinas por incrementos significativos del colesterol total a expensas del cLDL: 355 mg/dl. Su respuesta fue progresivamente satisfactoria aunque no de la manera que se esperaba, su cambio a un fibrato de segunda generación dio mejores resultados. La respuesta pobre al uso de estatinas como en este caso puede ser debido a un fenotipo de Apo E denominado alelo E2. Imagen 146-.- Hipercolesterolemia familiar (fenotipo IIa). Adviértase grandes tumores subcutáneos adherentes a los tendones de inserción de las falanges proximales (nudillos). Imagen 147.- Hipercolesterolemia familiar (fenotipo IIa). En detalles dos tumoraciones de aspecto nodular, no supurativos, ubicadas en el codo, que corresponden a xantomas tuberosos. Las imágenes 3 y 4 corresponden al mismo paciente. Imágenes 148.- Hipercolesterolemia familiar (fenotipo IIa). Tumoraciones de varios tamaños ubicadas en los codos sin signos de flogosis, e indoloras, sugestivas de xantomas tuberosos.Imágenes 149-150-151-152 .- Hipercolesterolemia familiar (fenotipo IIa). Paciente de sexo femenino de 17 años de edad que refiere lesiones de aspecto nodular de varios tamaños ubicadas en las manos preferentemente a nivel periarticular falángicas de ambas manos, no tienen aspecto inflamatorio y la piel que los recubre es normal, no refiere dolor. En ambos codos presenta nódulos de iguales características aunque son más grandes y de aspecto pediculado, muy sugestivos de xantomas tuberosos; refiere la paciente que le aparecen a partir del año de edad, estos han venido aumentando tanto tamaño y otros de nueva aparición (Colaboración de la Dra. Micaela López Obando, Bolivia).
INFORMACIÓN BÁSICA.- HIPERCOLESTEROLEMIAS FAMILIARES: Hipercolesterolemias familiares (HFs).- Dentro de las mas de 6 formas clínicas de HFs la forma heterocigota no es considerada una enfermedad rara en Europa (OMIM.143890-144010-602247-603813/ORPHA:-/CIE-10:E78.0).– No así la forma homocigota que si lo es en todo el mundo ( OMIM: /ORPHAN:391665 /CIE-10:E78.0).- La HF es una dislipidemia hereditaria caracterizada por una elevación permanente y aislada de las LDL circulantes.
Antiguamente denominada hipercolesterolemia familiar xantomatosa, así como con otras denominaciones: hiperbetalipoproteínemia familiar, xantomatosis hereditaria, xantomatosis familiar, xantoma tuberoso hereditario múltiple, xantelasma familiar, hipercolesterolemia familiar esencial. Como quiera que la enfermedad se defina, esta está determinada genéticamente, constituyendo quizás una equivocación el empleo del adjetivo familiar en lugar del de hereditario.
La HF en sus forma de presentación más común, la HF por déficit parcial (heterocigota), o total de receptores de LDL (homocigota), es una enfermedad hereditaria de transmisión autosómica dominante, conocida también como hiperbetalipoproteinemia, debido al aumento en la circulación de la fracción beta lipoproteína o LDL. La forma homocigota (HFHo) es muy rara (1/1 millón), y los individuos afectados carecen de RLDL, al tener mutado ambos alelos del gen, presentando concentraciones muy elevadas de colesterol plasmático total (entre 700 y 1000 mg/dl), y de entre 600-1.000 mg/dl de LDL con aparición durante los dos primeros años de vida y caracterizada por: depósitos cutáneos extravasculares de colesterol (xantomas cutáneos y/o tendinosos), niveles de LDL > 600mg/dl y arteriopatía (estenosis aórtica, EAC prematura) que se manifiesta antes de los 10 años de edad. Desarrollan aterosclerosis en una etapa temprana de la vida y a pesar de la instauración de tratamientos agresivos, los niveles elevados de LDL se modifican muy poco, falleciendo generalmente por EAC antes de los 30 años de edad. En la HF en su forma heterocigota (He) es mucho más frecuente y afecta aproximadamente a uno de cada 500 individuos. En ellos, el número de RLDL se reduce a un 50%, siendo suficientes los restantes para que se una la misma cantidad de LDL a la célula, pero a costa de elevarse de 2 a 3 veces la concentración plasmática de LDL. Esto hace que estos pacientes presenten un riesgo elevado de EAC precoz, entre los 30 y los 50 años, aunque muchos de ellos tienen una vida de duración normal.
El defecto básico de la HF radica en el RLDL, el cual es codificado por un gen de aproximadamente 45 kilobases (Kb), localizado en el brazo corto del cromosoma 19 (entre las regiones p13.1-p13.3) y consta de 18 exones y 17 intrones. Hasta enero de 2006 se habían descrito 861 mutaciones que afectan al gen que codifica a este receptor. Entre ellas destacan deleciones de distinto tamaño, originando algunas una proteína truncada, en tanto que las que afectan al promotor del gen impiden que éste se transcriba, no produciéndose por tanto la síntesis de la proteína correspondiente. Otras mutaciones incluyen sustituciones, y las que afectan al dominio citoplasmático del receptor, impiden su internalización. Las mutaciones del gen del RLDL causantes de la HF se suelen dividir en 5 clases (véase la Figura 8).
En la HF no solo preocupan las mutaciones presentadas en el gen del RLDL. Recientemente se ha observado que otros genes (APOB-100DF, PCSK9 y LDLRAP1/HAR) tienen participación directa en el desarrollo de la patología.
Herencia.- La HF de herencia autosómica recesiva (HAR/menos de 20 casos descritos hasta el momento) se caracteriza por XTs y/o aterosclerosis en niños con hipercolesterolemia grave, nacidos de padres con niveles lipídicos normales.
Las otras formas clínicas de HF, todas ellas autosómicas recesivas descritas son: la APOB-100DF. El gen que codifica para la apolipoproteína B-100 se encuentra en el brazo corto del cromosoma 2 (2p23).
La prevalencia en población europea se estima en 1 caso por cada 1.000 habitantes. Se han descrito 4 mutaciones en este gen (R3500Q27, R3500W, 28 R3531C, 29 y N3516K.30), con cuadros clínicos menos severos que los observados en pacientes con mutaciones RLDL. La HFAD de tipo 3, por mutacion en el gen PCSK9. El gen PCSK9, localizado en la región 1p34.1- p32.8, 32,33 codifica una proteína convertasa, tipo subtilisina/kexina. Se han publicado 4 mutaciones (Ser124arg,9 Asn157Lys,38 Phe216Leu,9 Asp374Tyr.39) causantes de un cuadro clínico indistinguible de la HF. Por consiguiente al tenor de lo enunciado anteriormente, la HF se caracterizada por el acumulo en sangre del cLDL o b-lipoproteína, y consecuentemente del colesterol en el plasma, por un defecto cuantitativo o cualitativo de los receptores de membrana para LDL. Las LDL son las principales transportadoras del colesterol en el plasma, como consecuencia de una disminución de su catabolismo.
Defectos moleculares.-El defecto básico consiste en la ausencia de receptores para LDL de forma total (fenotipo homocigoto) o parcial (fenotipo heterocigoto). Por ende se observa una severa insuficiencia de la depuración plasmática de lipoproteínas remanentes de VLDL o sea de las IDL y de LDL.
Esto conduce a la acumulación de partículas de cLDL en la sangre como resultado de la mayor conversión de IDL ® LDL, al igual que la menor depuración periférica de ésta última lipoproteína. Como resultado de ello, las células no pueden llevar a cabo los procesos normales de reconocimiento, captación, internalización o catabolismo de las LDL, elevándose las concentraciones del colesterol en el plasma. La HF constituye un claro ejemplo del importante papel que representa la elevación del colesterol plasmático en la génesis de la aterosclerosis.
Aunque la clasificación de la HF en fenotipo IIa y IIb es empírica, provisionalmente los términos tipo IIa y tipo IIb pueden ser útiles para expresarse en forma más abreviada, sea cual fuere el sentido que se quiera dar a la palabra. Se trata más bien de una anormalidad bioquímica común a varios desarreglos del metabolismo lipoproteico.
En los años ochenta, fue Goldstein y Brown demostraron el defecto básico de esta patología, con una descripción elegante y magistral, reconociendo que la alteración básica era en la ausencia o disminución de la actividad del RLDL, como resultado de varias mutaciones del gen que codifica la síntesis de estos receptores, localizado en la zona distal del brazo corto del cromosoma 19. Las mutaciones en el locus para el RLDL origina cierta incapacidad para producir el precursor de dicho receptor, o bien para producir un receptor anormal que no llega a la forma final de un RLDL, por último, se produce un receptor pero defectuoso. La acumulación de cLDL en el plasma resulta por consiguiente, de una reducción significativa de la capacidad de catabolizar partículas por vía del RLDL. Pese a que la presentación fenotípica de la HF sea bastante similar en los diferentes pacientes, la naturaleza precisa del defecto genético puede variar considerablemente.
La mayoría de los pacientes con HFHo confirmada genéticamente tienen 2 alelos mutados del gen RLDL (>95%; OMIM 606945) y cada uno de sus progenitores presenta HFHe. Recientemente se han identificado mutaciones en alelos de otros 3 genes como causantes de casos con un fenotipo grave de HFHo. Estos genes secundarios son APOB (2-5%; OMIM 107730), que codifica la apolipoproteína (apo) B; PCSK9 (<1%; OMIM 607786), que codifica la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9), y LDLRAP1 (<1%; OMIM 695747), que codificala una proteína adaptadora 1 del RLDL, que causa un fenotipo recesivo (HAR), ya que los progenitores portadores presentan perfiles lipídicos normales. Los pacientes son homocigotos, con la misma mutación en ambos alelos del mismo gen o, más a menudo, heterocigotos compuestos con mutaciones diferentes en cada alelo del mismo gen, o heterocigotos dobles con mutaciones en 2 genes diferentes que afectan a la función del RLDL.
Figura 7.- Receptor de LDL. LDLRAP1 sirve como una proteína adaptadora del RLDL en el citoplasma en colaboración con clatrina AP-2 y Dab2. Este complejo se internaliza en los hepatocitos. PCSK9 es un modulador de la endocitosis hepática que degrada el RLDL. Brown y Goldstein pasaron a caracterizar la llamada «vía receptor LDL’, revelando detalles de la endocitosis mediada por receptores (RLDL) que no sólo había implicaciones para otras rutas de largo alcance, pero serían posteriormente permitir la identificación de otros defectos genéticos que causan el mal funcionamiento del RLDL. Se clasifican los defectos celulares de la función del receptor de LDL en cinco grupos:-ligando de unión defectuosa; transportar defectuosa; internalización defectuosa; reciclaje defectuosa; y ‘nulo’, que dio lugar a ninguna proteína (RECEPTOR) detectable. La mayoría de las mutaciones del gen RLDL se producen en el dominio de unión a ligando (exón 2-4) y la homología precursor EGF (exones 7-14). La estructura básica del gen RLDLR y áreas de mutación común se muestra a continuación, en la figura 8.
Antecedentes familiares.-Es esencial una evaluación cuidadosa de los antecedentes familiares para la valoración exhaustiva de una posible HF en general y de una HFHo en particular. En el caso de las mutaciones autosómicas dominantes (en genes RLDL, PCSK9 y APOB), ambos progenitores son heterocigotos obligados y, por tanto, muestran niveles elevados de cLDL (frecuentemente percentil >95 según los criterios de edad y sexo específicos del país) y antecedentes familiares positivos de ECVA prematura (<55 años en varones y < 60 años en mujeres entre familiares de primer grado). En el caso de la HAR (debida a mutaciones en LDLRAP1), los progenitores pueden presentar niveles de cLDL en el intervalo normal, y la determinación de un árbol genealógico extenso de la familia puede revelar un patrón autosómico recesivo heredado. El cribado en cascada familiar ofrece prospectivamente a los padres con HFHe la posibilidad de consejo genético e identificar a pacientes con HFHo al nacer, permitiendo así el inicio precoz del tratamiento. La identificación de la HFHo también puede guiar la detección en cascada «inversa» para progenitores y familiares a fin de identificar a los pacientes con HF.
Prevalencia.- Los individuos con HF sólo representan un pequeño porcentaje de la población con hipercolesterolemia primaria. Es así, que sólo alrededor de 1:20 individuos de la población mundial con incrementos del CT plasmático con un patrón de hiperlipoproteinemia de tipo IIa presentan HF.
Figura 8.- Mutaciones del gen del Receptor de LDL- Las mutaciones del gen rLDL se dividen en 5 clases en función de su efecto sobre el ciclo del RLDL. Mutaciones de clase 1 (alelos nulos): son las más graves, dado que el defecto conlleva la ausencia total de producción del receptor. Mutaciones de clase 2 (alelos defectuosos para el transporte): es la más frecuente y se debe al bloqueo en el proceso de transporte del receptor sintetizado desde el retículo endoplásmico hasta el aparato de Golgi; la mayoría de estas mutaciones se localizan en los exones que codifican el dominio de unión al ligando o en el dominio homólogo al precursor del factor de crecimiento endotelial (EGF). Mutaciones de clase 3 (alelos defectuosos para la unión): imposibilidad de unión de LDL al receptor celular. Mutaciones de clase 4 (alelos defectuosos para la internalización): no transportan las LDL hacia el interior de la célula. Mutaciones de clase 5 (alelos defectuosos para el reciclado): impiden que los receptores LDL internalizados regresen de nuevo a la superficie celular para iniciar de nuevo el proceso de captación del cLDL. Fuente: Real JT, Carmena R. Estudio Genético de las hiperlipemias. JANO EMC 1999 Feb; 56 (1286): 64.
La HF es la enfermedad monogénica más frecuente de la especie humana y de la que mejor se conoce de su comportamiento molecular. Para la HF en su forma heterocigota, los estudios de prevalencia denuncian una incidencia de al menos 1 por cada 500 habitantes en Europa, Japón y EE.UU. de Norteamérica, aunque en ciertas zonas de Canadá (Quebec) la incidencia es mucho más elevada, 1 por 122 habitantes. No es raro observar que esta incidencia aumente significativamente en ciertas poblaciones como la sudafricana -Ciudad del Cabo-, situándose en 1 por cada 71 habitantes, igual cosa ocurre con la población libanesa. Sin embargo, en poblaciones endogámicas la prevalencia es mucho más alta.
La HF en su forma homocigota, muy rara en Europa central -aproximadamente 1-5:10.000.000 de habitantes-. En Suiza sólo se ha reportado un caso. Siendo la incidencia en el resto de países de 1/1.000.000 de habitantes, sin embargo, datos recientes sugieren que esta cifra puede estar entre 1 por cada 300.000 a 160.000 habitantes, o incluso puede ser mayor en poblaciones con características especiales o con mayor frecuencia de consanguinidad. Para la HFHe 1:500; para la APB-100DF 1:1.000; para la HF de Tipo 3 (PCSK9) < 1:2.500 y para la HAR <1:5.000.000 habitantes.
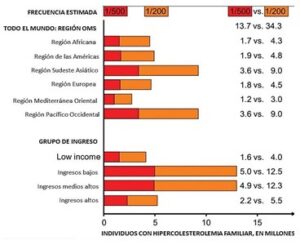
Figura 9.- Estimados en millones de personas en todo el mundo con HF por regiones de la OMS y por grupos de ingresos. Las estimaciones se muestran con la frecuencia teórica de la HFHe de 1:500 en la población general, asi como para la frecuencia detectada directamente 1:200 en población danesa, un país típico del norte de europa. Fuente: Børge G. Nordestgaard M. John Chapman Steve E. Humphries Henry N. Ginsberg Luis Masana Olivier S. Descamps Olov Wiklund Robert A. Hegele Frederick J. Raal Joep C. Defesche. La hipercolesterolemia familiar está mal diagnosticada y no se trata en la población general: guía para los médicos clínicos para prevenir la enfermedad coronaria: Declaración de consenso de la Sociedad Europea de Aterosclerosis. European Heart Journal, volumen 34, Número 45, 1 de diciembre de 2013, páginas 3478 3490, https://doi.org/10.1093/eurheartj/eht273
En nuestro país, describo por primera vez esta entidad en el año de 1985 en una paciente originaría de la Provincia Peninsular de Santa Elena; debido a la rareza de la misma es motivo de presentación de algunas fotografías (imagen 130) de las manifestaciones cutáneas, que presentaré más adelante. Es de anotar que la paciente no respondió a ningún tipo de terapia, falleciendo a los 25 años de edad por IAM.
Figura 10 .- Causas de Hipercolesterolemias Familiares: Más del 80% corresponde a la HF por ausencia del RLDL. Un 5% corresponde a ApoB100 Defectuosa Familiar, y <1% a la HF por mutación del gen de la PCSK-9. Las otras formas de HFs se desconocen sus porcentajes de responsabilidad.
Distribución.- Existen grandes variaciones en cuanto a la presentación de la HF en todo el mundo, quizá en parte debido a la consanguinidad. Hay frecuencias anormales en Líbano y Sudáfrica, donde se observa en Transval un caso de homocigotos por cada 30.000 habitantes. En relación con el estado heterocigoto, en el Líbano la frecuencia de esta forma puede alcanzar hasta 1:80.
Del 20 al 50% de todas las hiperlipoproteinemias primarias existentes corresponden a los fenotipos IIa y IIb. Un 10% de los heterocigotos para HF pueden expresar un fenotipo IIb.
Por último, la HFHe, representan aproximadamente 5-10% de los adultos jóvenes internados con IAM.
Figura 11.- Segregación de un gen autosómico dominante. Cuando uno de los progenitores son heterocigotos afectados para la HF. La HF se transmite de forma autosómica dominante, esto quiere decir que una persona con HF tiene el 50% de probabilidades de transmitir el gen anormal a su descendencia. Por lo tanto, aproximadamente la mitad de los hijos, independientemente del sexo, padecerán también de HF.
Herencia.-La HF se hereda de forma autosómica dominante y tiene una penetrancia cercana a 100%. Existen dos formas de HFs, la HFHe, cuando se hereda sólo un alelo mutado de uno de los progenitores, y la de HFHo, cuando se hereda de ambos progenitores la misma mutación. La HF compuesta se produce cuando se hereda una mutación distinta en el mismo gen de cada progenitor y tiene una expresión clínica similar a la HFHo.
La forma de HFHe tiene mejor pronóstico en el contexto de la HF. Las mujeres con HF parecen tener un mejor pronóstico que los varones. En la figura 11 se resume la transmisión vertical y las probabilidades de herencia monogénica de la HF.
Los fenotipos IIa y IIb pueden encontrarse indistintamente en los miembros de una misma familia para la HF, y esta discordancia de las lipoproteínas plasmáticas puede presentarse en una misma o en distintas generaciones.
Patogenia.- El fenotipo que nos ocupa, está determinado genéticamente por una o dos mutaciones para el gen que codifica la síntesis del RLDL (Apo-B100: E), conduce a un defecto, parcial o total, en la actividad de los RLDL que regulan el aclaramiento plasmático de las LDL y la síntesis del colesterol intracelular.
La HF es una enfermedad grave compleja e infradiagnosticada. Más de 1.700 mutaciones genéticas asociadas han sido identificadas.
La HF está causada por mutaciones que resultan en un defecto en la endocitosis de LDLs. Para las formas dominantes, se han identificado mutaciones en los genes: RLDL(responsable de entre los 2/3 y 3/4 de los casos con herencia dominante), gen APOB, que codifica para el receptor ligando LDL y gen PCSK9 (causante del 1-3% de las HFs), un modulador de la endocitosis hepática. Las mutaciones responsables de las formas recesivas se han identificado en los genes LDLRAP1 y ABCG5/ABCG8.
Características Clínicas.- Las manifestaciones clínicas clásicas de la HF están integradas en una tétrada compuesta: hipercolesterolemia, xantomas tendinosos – cutáneos y aterosclerosis anticipada y acelerada. Sin embargo, como dato clínico más característico es la detección de valores elevados de CT y de cLDL. La hipercolesterolemia se manifiesta desde el nacimiento y en ambos sexos, incluso pudiendo detectarse ya en la sangre del cordón umbilical, lo que permite el diagnóstico prenatal tanto en homocigotos como en heterocigotos para el trastorno.
En la forma de HFHe, las concentraciones plasmáticas de CT en los primeros meses de vida se sitúan alrededor de 300 mg/dl, y en la edad adulta con valores entre 350 y 500 mg/dl. De igual manera los niveles de cLDL se encuentran por regla general >190 mg/dl.; pero este diagnóstico es también muy probable en personas adultas menores de 16 años de edad, con valores de CT >260 mg/dl.
Los heterocigotos suelen mantenerse asintomáticos hasta que presentan un evento vascular coronario, por lo general a partir de los 40 años de edad en los varones y de los 50 años en las mujeres.
En el raro estado de la HFHo la concentración del CT suele ser superior a 600 mg/d en la infancia, superando incluso los 1.000 a 5.000 mg/dl en la edad adulta.
En los pacientes adultos son característicos, el AC del tipo senil prematuro (<45 años de edad) y xantelasmas (imagen 38), a menudo grotescos, así como XTs en el tendón de Aquiles (imagen 141), el dorso de las manos -particularmente en el pliegue entre el pulgar y el índice, antebrazos (imagen), codos, rodillas y grandes pliegues de los glúteos; también pueden presentarse en otras zonas como la fascia plantar y debajo del periostio. La presencia de XTs valvulares de preferencia en la válvula aórtica dan lugar a estenosis precoz ya desde la niñez o la juventud, produciéndose en éstas verdaderas estrecheces, acompañándose de un soplo de estenosis, dificultando aún más la función cardíaca de por sí comprometida en estos pacientes. El 31-50% de los pacientes suelen presentar un AC prematuro.
La prevalencia de xantomatosis en los pacientes con HFHe anglosajones y escandinavos puede llegar hasta el 75%. Esta prevalencia llega casi al 100% en los pacientes con HFHo. Estos últimos presentan algún tipo de XT cutáneo (tuberosos y/o tendinosos, planos) y de órganos internos en los primeros 5 años de vida, incluso a veces al nacer (claves para el diagnóstico).
La prevalencia estimada de XTs en la HFHo no ha sido sistemáticamente establecida, destacando que la presencia de XTs dentro de la primera década de la vida, son patognomónicos de la HFHo.
Al contrario, en la HFHe, la frecuencia de presentación va aumentando con la edad, de tal manera que a la edad de 40 años presentan esta alteración el 75-80% de los mismos, siendo el primer signo la presencia de XTs tendinosos en el tendón de Aquiles.
Las lesiones cutáneas pueden ser tuberosas o planas. Las lesiones tuberosas se presentan en heterocigotos y homocigotos, con mayor frecuencia sobre los codos y talones. Las lesiones planas, que son ligeramente elevadas, se hallan mucho más a menudo en homocigotos que en heterocigotos y tienden a distribuirse por las nalgas, muslos, codos rodillas y yemas de los dedos. Los XTs planos de la piel periocular se denominan XTs; estas lesiones se producen en heterocigotos y homocigotos, pero también pueden presentarse en adultos no lipémicos. Es interesante que estos pacientes a futuro desarrollen hiperlipidemia. He observado que estos pacientes presentan mayor nivel de apoB. Los XTs tendinosos se encuentran en ambos genotipos de HF, con máxima frecuencia en los tendones de Aquiles y en los tendones extensores de las manos. En éstas las lesiones tendinosas provocan dolor, distorsión y erosión en las articulaciones digitales. Puede haber XTs subperiósticos bajo las rodillas y en las apófisis olecranianas, en ambos genotipos. Salvo los XTs tendinosos que a veces se encuentran en los adolescentes afectados, estos signos rara vez se encuentran en los niños con HFHe. Por tanto, dada su rareza en los niños sanos, la tendinitis aquílea en un adolescente debe hacer sospechar el diagnóstico de HF. Contrario a estos hallazgos, Alonso-Villaverde y cols. Han publicado recientemente datos sobre la escasa frecuencia de XTs en enfermos españoles con HFHe.
Diagnóstico clínico de la HF en adultos.- El diagnóstico de la HF se basa en niveles elevados de cLDL (generalmente >220 mg/dL), historia familiar de hipercolesterolemia (especialmente si hay niños o adolescentes afectos), presencia de EAC prematura y depósitos de colesterol en forma de XTs y/o AC precoz. Los XTs tendinosos son patognomónicos de HF; sin embargo, se encuentran en menos del 30% de los casos confirmados de HF. Su ausencia no excluye el diagnóstico de HF. Los datos de sospecha de la HF se consignan en la tabla 6.
La obtención del árbol familiar es esencial para evaluar la probabilidad de HF y para posteriormente realizar la detección familiar. Existen tres herramientas diferentes para establecer el diagnóstico clínico de la HF en el caso índice (CI): el programa MedPed, el Simon Broome británico y los criterios de la Red de Clínicasde Lípidos Holandesa (RCLH). Se basan en un sistema de puntuación, según la historia personal y familiar de determinadas variables (tabla 6).
El diagnóstico clínico es de certeza cuando la puntuación es ≥8 y de probabilidad cuando es ≥6. La precisión de los criterios clínicos se ha comparado con el diagnóstico genético que es el gold standard, siendo los criterios de la RCLH los que en conjunto tienen mejor sensibilidad y especificidad. Los criterios diagnósticos de la RCLH solo se deben utilizar para el diagnóstico del CI mayor de 18 años y nunca en sus familiares.
En casos excepcionales, los pacientes pueden presentar XTs ectópicos gigantes en el cerebro, mediastino y músculos de los glúteos. La presencia de niveles marcadamente elevados de cLDL y la ausencia de síntomas neurológicos, cognitivos y oftálmicos en pacientes con HFHo los distingue de pacientes con XCT.
Criterios diagnósticos de hipercolesterolemia familiar heterocigota (Tabla III)
Diagnóstico molecular de la HF.-La HFHo es un desorden autosómico dominante. A lo largo de la historia se ha reportado una prevalencia de 1 por cada 1.000.000 habitantes; sin embargo, datos recientes sugieren que esta cifra puede estar entre 1 por cada 300.000 a 160.000 habitantes, o incluso puede ser mayor en poblaciones con características especiales o con mayor frecuencia de consanguinidad.
En sí, la HF en todas sus variantes clínicas, es causada principalmente por la mutación de ambos alelos del gen del RLDLR (90%) y, con menor frecuencia, por mutación de apolipoproteina B100 (5%), o en la proproteína convertasa subtilisina/kexina tipo 9 (PCSK-9) o proteína adaptadora del RLDL (1%).
Diagnóstico analítico de la HF.-
El estudio analítico debe efectuarse en las siguientes condiciones:
1. Ayuno de 12 horas.
2. Sin modificaciones de la dieta habitual.
3. Libre de enfermedad desde varias semanas antes.
4. Teniendo en cuenta la toma de ciertos fármacos que pueden modificar los lípidos.
En caso de valores lipídicos alterados, se repetirá el estudio analítico tres o cuatro semanas después, para confirmar la alteración de la analítica.
Se deberá realizar una historia familiar (antecedentes de ECV prematura en padres o abuelos, antecedentes en padre o madre de dislipemia genética). Asimismo, conocer los antecedentes personales: hábitos alimentarios, estilos de vida, toma de fármacos, hábito tabáquico y se descartarán posibles causas de dislipemia secundaria.
En la exploración física, se anotarán las medidas antropométricas: peso, talla, IMC y estadio puberal, además de la toma de la tensión arterial.
En el estudio analítico, se solicitará:
• Hemograma, Bioquímica (perfil hepático y renal) y hormonas tiroideas.
• Perfil lipídico en ayunas: CT, TG y lipoproteínas (C-LDL, C-HDL y C-VLDL).
• Según sospecha etiológica: apolipoproteína A, B, y Lp(a).
El objetivo principal de diagnosticar una hipercolesterolemia en la infancia es el inicio de un tratamiento eficaz, especialmente la instauración de hábitos de vida saludables lo antes posible, para prevenir el desarrollo de ECV que se manifiesta en la edad adulta.
Cribado de las dislipemias
La práctica del cribado de las dislipemias en niños o adolescentes es un tema controvertido, en lo que respecta a quién y cuándo hacerlo, debido a los riesgos desconocidos de un tratamiento hipolipemiante a largo plazo iniciado en edades tempranas. Existen diferentes estrategias para la detección en los niños, como son: el cribado en cascada familiar, el cribado universal y el cribado selectivo basado en la historia familiar. En España y en otros países europeos, el cribado en los niños se suele hacer como parte del cribado en cascada familiar, cuando se conoce el diagnóstico en la familia. El cribado selectivo debe realizarse a la población infanto-juvenil de riesgo entre los 2 y los 8 años, y entre los 12 y los 16 años(5). Se considera población de riesgo, si existe historia familiar positiva o factores de riesgo en el niño:
Historia familiar
ECV prematura en padres, abuelos o tíos (varones < 55 años o mujeres < 65 años).
Padres con dislipemia conocida o CT mayor o igual a 240 mg/dl.
Factores de alto riesgo
Hipertensión arterial que requiere tratamiento farmacológico (TA > P 99 + 5 mmHg).
Hábito tabáquico.
IMC ≥ percentil 97.
Presencia de condiciones de alto riesgo: diabetes mellitus, enfermedad renal crónica, trasplante de órganos, enfermedad de Kawasaki con aneurismas.
Factores de riesgo moderado
Hipertensión arterial que no requiere tratamiento farmacológico.
IMC ≥ percentil 95 y < percentil 97.
C-HDL< 40 mg/dl.
Presencia de condiciones de riesgo moderado: enfermedad de Kawasaki con aneurismas en regresión, enfermedad inflamatoria crónica (lupus eritematoso, artritis idiopática juvenil), VIH y síndrome nefrótico.
Diagnóstico.- El estudio analítico debe efectuarse en las siguientes condiciones:
1. Ayuno de 12 horas.
2. Sin modificaciones de la dieta habitual.
3. Libre de enfermedad desde varias semanas antes.
4. Teniendo en cuenta la toma de ciertos fármacos que pueden modificar los lípidos.
En caso de valores lipídicos alterados, se repetirá el estudio analítico tres o cuatro semanas después, para confirmar la alteración de la analítica.
Se deberá realizar una historia familiar (antecedentes de ECV prematura en padres o abuelos, antecedentes en padre o madre de dislipemia genética). Asimismo, conocer los antecedentes personales: hábitos alimentarios, estilos de vida, toma de fármacos, hábito tabáquico y se descartarán posibles causas de dislipemia secundaria.
En la exploración física, se anotarán las medidas antropométricas: peso, talla, IMC y estadio puberal, además de la toma de la tensión arterial.
En el estudio analítico, se solicitará:
Hemograma, Bioquímica (perfil hepático y renal) y hormonas tiroideas.
Perfil lipídico en ayunas: CT, TG y lipoproteínas (C-LDL, C-HDL y C-VLDL).
Según sospecha etiológica: apolipoproteína A, B, y Lp(a).
Confirmar la trasmisión vertical de la hipercolesterolemia y/o ECV prematura en uno de los padres.
Niveles de C-LDL ≥ 190 mg/dl o bien niveles de C-LDL ≥ 150 mg/dl, cuando se tiene la confirmación genética de HF en uno de los padres.
El estudio genético nos permitirá dar una información pronóstica a los pacientes y se realizará mediante:
– La demostración de mutaciones en el gen del receptor LDL (LDLR, 19p13.2). La tasa de detección de una mutación funcional en casos con diagnóstico clínico varía del 20 al 80%, por tanto, un test genético negativo no excluye el diagnóstico, sobre todo, cuando el fenotipo sugiere una HF.
– Otras causas menos frecuentes son las mutaciones en el gen de la ApoB (apoB100; 2p24) y en el gen de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9, 1p34.1-p32)(14).
– Como resultado de estos defectos moleculares se produce una disminución en la eliminación del colesterol plasmático unido a lipoproteínas de baja densidad, y esto conlleva a que estos pacientes presenten desde el nacimiento valores plasmáticos elevados de CT y C-LDL.
Diagnóstico diferencial.- El diagnóstico diferencial de la HF esta en relación con su genotipo. El diagnóstico diferencial de la HFHo incluye la HLFC, la DBLPF (Fenotipo III), xantomatosis cerebrotendinosa, sitosterolemia e HFHe
Aunque las características de cada patología pueden sugerir el diagnóstico clínico, en muchos casos es necesaria la realización de pruebas moleculares para confirmarlo. La forma de HFHe se debe realizar con la HLFC (tabla 11) y con la HP con agregación familiar (caracterizadas por hiperlipidemia LDL moderada fluctuante, normalizada con una dieta controlada en lípidos, sin historial familiar de HF a lo largo de tres generaciones). Los marcadores diagnósticos permiten distinguir clínicamente las formas adultas o infantiles de la HFHe de las formas combinadas. Al igual que la HFHo el diagnóstico se confirma mediante genética molecular.
Debido al alto riesgo de ECV, el diagnóstico de HF en un individuo, debe conducir a la exploración y manejo de todo el nucleo familiar. El cribaje familiar debe comenzar tan pronto como sea posible, preferiblemente durante la fase de latencia de la enfermedad, cuando las manifestaciones arteriales son reversibles. El diagnóstico prenatal puede proponerse a las familias que presenten al menos un caso de HFHo, y las que las mutaciones responsables parentales hayan sido identificadas.
Los XTs tendinosos en especial del tendón de Aquiles es una característica de la HF, pero también pueden observarse en los sujetos normocolesterolémicos con xantomatosis cerebrotendinosa (Enfermedad de Van Bogaert), y con HF seudohomocigótica como la sitosterolemia; (ver diagnóstico diferencial), o enfermedades linfoproliferativas. El 80% de los sujetos adultos con HFhe tiene XTs tendinosos, aumentando su incidencia hasta más del 90% en la quinta década.
El lípido principal de las lesiones xantomatosas de la HF es el colesterol libre y esterificado, pero también se encuentran fosfolípidos y TGs. Se supone que el colesterol de estas lesiones se origina del plasma más que por síntesis local.
Es importante diferenciarla de otras causas de hipercolesterolemias secundarias como: el hipotiroidismo, el SN, la colestasis y el tratamiento con determinados fármacos (esteroides, inmunosupresores, etc.).
Las lesiones ateroscleróticas de los vasos y la de los XTs tendinosos son histológicamente similares. Además, ambas aumentan con la edad y se estabilizan o regresan parcialmente con el tratamiento hipolipemiante agresivo, lo que indica una asociación directa entre el depósito lipídico extravascular y la gravedad de las lesiones coronarias en la HF.
Los XTs son hiperplasias simples del sistema monocítico-fagocitario. Consideradas además, como reacciones locales celulares frente al trastorno metabólico lipídico. Las células de XTs o células espumosas, son macrófagos que por su capacidad fagocítica, se llenan de gotitas de lípidos. En los cortes histológicos, el citoplasma es reticulado o espumoso, por que el procesamiento automático disuelve y extrae los lípidos. En las muestras congeladas o fijadas en formalina y coloreadas con rojo escarlata o Sudán, las gotitas lipídicas se aprecian con claridad. En general el núcleo de la célula de XT es único, pero puede ser múltiple, en este caso, la distribución es irregular, como en las células gigantes de cuerpo extraño, o se forma una corona alrededor de un islote de citoplasma no espumoso, rodeado por citoplasma espumoso; estas últimas se llaman células gigantes de Touton.
Los XTs típicos que se presentan en el fenotipo que nos ocupa, son los XTs tuberosos, estos se presentan como grandes pápulas, tubérculos o nódulos que se unen formando placas como elemento característico más o menos irregulares, de color amarillo-naranja, localizados sobre todo en áreas de extensión y de micro traumatismos -codos, rodillas, nalgas- y en las manos, siguiendo los surcos de las palmas y dedos.
Los XTs cutáneos en las HFs suelen aparecen con cifras >400 mg/dl de colesterol plasmático (en la sitosterolemia con valores muchos más bajos), y representa el primer signo detectado por familiares de los niños homocigotos para HF. Los XTs son asintomáticos, la evolución es larga y son rebeldes al tratamiento, no obstante, he observado reducciones significativas del tamaño de los XTs usando el desaparecido probucol.
En aproximadamente el 50% de homocigotos se producen ataques transitorios de poliartritis hiperlipoproteinémica, que duran unos pocos días, de etiología poco clara, de presentación aguda, y a menudo migratorias, afectando a grandes articulaciones periféricas, éstas son afectadas por dolor, que oscila desde artralgías moderadas hasta molestias intensas, acompañadas de tumefacción sinovial, eritema y dolor a la presión.
La duración de la afectación de una articulación concreta oscila entre unos pocos días y un mes, y los síntomas emigran de una articulación a otra, lo que puede inducir al diagnóstico erróneo de fiebre reumática. La artritis puede afectar rodillas, tobillos, cadera, codos, muñecas y también las pequeñas articulaciones de las manos.
Radiológicamente las articulaciones son normales. Puede haber erosiones de las falanges que no comprometen a la superficie articular. Se han comunicado casos anecdóticos de XTs en el SNC que simulaban tumores neurológicos primarios Se ha descrito de igual manera una miopatía (mialgias, calambres musculares, intolerancia al ejercicio y ligera paresia proximal) lipídica asociada a la HFHe. Un 10% de los pacientes con HF pueden presentar pérdida neurosensorial de la audición, que podría se progresiva, fluctuante o súbita.
Diagnóstico diferencial con la sitosterolemia.- Aunque en la mayoría de los casos el diagnóstico de HFHo es relativamente sencillo, otro trastorno del metabolismo lipídico, la sitosterolemia (también llamada fitosterolemia), puede tener una presentación clínica similar, con la presencia de XTs tendinosos y/o tuberosos en niños asociada a un notable aumento de los niveles de colesterol en plasma (aunque esto es discutible y variable) y complicaciones arterioscleróticas prematuras. Sin embargo, es relevante que la enfermedad arteriosclerótica no siempre esté presente en los sujetos sitosterolémicos definidos genéticamente, como se muestra en una reciente publicación. Al igual que la HAR, la sitosterolemia tiene un patrón de herencia autosómica recesiva, por lo que los progenitores pueden presentar niveles normales de colesterol. Dos características importantes diferencian la sitosterolemia de la HFHo:
a) concentraciones plasmáticas marcadamente elevadas (> 30 veces) de fitoesteroles, y b) niveles elevados de colesterol que responden bien a la dieta y a los secuestrantes de ácidos biliares o ezetimiba y que pueden desaparecer después de las 2 primeras décadas de vida.
El diagnóstico de la sistoterolemia se confirma mediante análisis genético, con mutaciones en 2 genes de los ATP binding cassette transporters, ABCG5 y/o ABCG8 (véase más adelante: Sistosterolemia). En resumen, el Grupo de Consenso de la Sociedad Española de Aterosclerosis (SEA) recomienda que el diagnóstico de la HFHo se realice mediante una cuidadosa evaluación de las características clínicas y de los antecedentes familiares, así como mediante pruebas genéticas, especialmentecuando el diagnóstico clínico de la HFHo sea dudoso o para facilitar el cribado en cascada familiar inversa. La detección en cascada familiar inversa se recomienda siempre en cualquier caso.
Pronóstico.- Se calcula que la HAD acorta la esperanza de vida, en más de 25 años, con respecto a la población general.
Factores ambientales que influyen sobre la variabilidadclínica.- El profundo trastorno metabólico existente en estos pacientes hace que la hipercolesterolemia sea su factor de riesgo fundamental. Por ello el tratamiento farmacológico hipolipemiante es hoy el eje principal de su manejo. Sin embargo, datos del Registro Español muestran que factores nutricionales, como es el sobrepeso, pueden ser claves como predictores de un mayor riesgo. Por ello la alimentación saludable puede ser un elemento clave en su manejo. Existen dos hechos fundamentales en torno al efecto preventivo de la dieta: la gran variabilidad de la respuesta individual y un nuevo paradigma, el que los nutrientes ejercen múltiples efectos biológicos, que van más allá de su beneficio sobre el colesterol.
Con respecto a los efectos pleiotrópicos de los nutrientes, es importante conocer si estos pacientes se benefician de una dieta sana (tipo mediterránea), no sólo por su efecto sobre el colesterol sino por su potencial efecto sobre otras variables predictivas, relacionadas con la dieta. Entre ellas se incluyen, el tamaño de la LDL, la oxidación de LPs, el metabolismo postprandial de TGs y factores hemostáticos.
El primer aspecto se enmarca dentro de la variabilidad dependiente de la interacción genético-ambiental en la expresión del colesterol plasmático, escasamente conocido en los pacientes con HF. Uno de los factores genéticos, sobre el que se está centrando el interés, es el de las variantes en los genes de los transportadores ABCG5 y ABCG8. Ambos se han implicado en la absorción de los esteroles de la dieta y en la regulación del transporte lipídico intracelular.
Se han propuesto distintos factores adicionales que condicionan el RCV, entre ellos la homocisteína. Así, el exceso de homocisteína es un FRCV independiente y tiene un efecto sinérgico con otros factores aterogénicos. La homocisteína favorece la lipoperoxidación y la trombosis e influye en los mecanismos que regulan la biosíntesis de colesterol. Las concentraciones homocisteína dependen de la ingesta de metionina, de la actividad de las enzimas que intervienen en su metabolismo y de las vitaminas que actúan como cofactores (ácido fólico, cobalamina y piridoxina). Se han identificado distintas variantes de los genes que codifican para estas enzimas relacionadas con concentraciones elevadas de homocisteína. Una de las más frecuentes es la mutación C677T del gen de la MTHFR, que se presenta con carácter homocigoto en un 13% de la población general.
Consideraciones adicionales en cuanto al diagnóstico: A Nivel mundial la mayoría de los pacientes con HF están sin diagnosticar y por lo tanto sin tratamiento, o bien con tratamiento insuficiente. El grado de infradiagnóstico y mal tratamiento de individuos en la población general con HF es en gran parte desconocido.
En general, se cree que entre los blancos, 1/500 son heterocigotos para FH y 1/1 000 000 son homocigotos; sin embargo, incluso estos individuos no se diagnostican en la mayoría de los países. Por otra parte, estas prevalencias probablemente representan subestimaciones y las ECVs como es la principal causa de muerte en el mundo. De hecho, muchas personas y familias con HF pueden simplemente pasar desapercibidas entre la gran cantidad de personas con EAC causada por factores de riesgo más comunes y, como consecuencia, estar infradiagnosticados y tratados de forma inadecuada para niveles de colesterol genéticamente elevados.
Consideraciones generales en cuanto al tratamiento de las hipercolesterolemias (exclusivamente para las formas de HP, la HLFC y formas secundarias). En estos pacientes la terapia recomendada es en primer lugar, Cambios Terapéuticos en los Estilos de Vida (CTEV) de vida, con una dieta equilibrada y si no se consigue mantener unos niveles adecuados de colesterol en un tiempo estimado, el tratamiento farmacológico recomendado son las estatinas.
Para iniciar estrategias de Prevención Primaria (PP) es prioritario, conocer el riesgo cardiovascular global antes de que aparezcan manifestaciones clínicas.
Cuando un paciente ha presentado síntomas clínicos cardiovasculares o tienen la enfermedad silente, debemos aplicar medidas de PS para evitar la aparición de nuevas manifestaciones clínicas. Por tanto, además del control de factores de riesgo, se necesitarán fármacos e incluso podría llegar a ser necesario el uso de intervenciones de revascularización, pero la actitud prioritaria sigue siendo la modificación de los CTEV hacia unos hábitos saludables.
Los grupos de intervención son:
Pacientes con enfermedad coronaria establecida
Personas sanas con riesgo alto de desarrollar enfermedad coronaria u aterosclerótica o sea, pacientes con cualquiera de las formas clínicas de HF.
Familiares de primer grado de pacientes con enfermedad coronaria o personas sanas con riesgo cardiovascular muy alto.
Dentro de la prevención y el tratamiento, éste ha ido evolucionando y cambiando a lo largo del tiempo, y hay que individualizar a cada paciente, ya que dependiendo de los datos de colesterol se tratara de una forma u otra.
En primer lugar, se intentará el tratamiento de aquellos pacientes sin valores muy elevados de colesterol con medidas saludables: que se basan en adquirir hábitos de vida saludables orientados a reducir el riesgo cardiovascular, en nuestro país no contamos con la dieta mediterránea atribuible a ella tantas evidencias de prevención cardiovascular; se pueden destacar:
– Dejar de fumar.
– Reducir el consumo de alcohol (ingesta moderada) a menos de 30g/día en hombres y 20g/día en mujeres. En casos de hipertrigliceridemia suprimirlo totalmente.
– Dieta hipocalórica reduciendo la ingesta de ácidos grasos saturados y trans ácidos grasos y colesterol por debajo de 300mg diarios, aumentando el consumo de hidratos de carbono hasta un 50-55% del aporte calórico, limitar el consumo de sal.
– Consumir pescado al menos 3 días por semana, sobre todo pescado azul.
– Alimentos ricos en fibra (25-30g/día)
– Tomar entre 1 y 5 raciones de frutos secos por semana.
– Ejercicio físico aeróbico y adaptado a las características del sujeto.
– Dieta hipolipemiante.
Si tras estas medidas durante un periodo de tres a seis meses persiste la dislipemia, será necesario recurrir al tratamiento farmacológico.
En cuanto al tratamiento farmacológico, las estatinas (inhibidores de la HMG-CoA reductasa) son los fármacos de elección, también tenemos los fibratos en especial los de segunda generación (Bezafibrato, Fenofibrato y Ciprofibrato) que reducen la síntesis y favorecen la eliminación biliar del colesterol, útiles en las dislipemias mixtas, con predominio de hipertrigliceridemia y las resinas que interfieren en la absorción de los ácidos biliares en el intestino aumentando el paso del colesterol hepático a ácidos grasos.
En los ensayos realizados en PP con estatinas, se ha observado una reducción de la morbimortalidad cardiovascular y de la mortalidad total, ya que son los hipolipemiantes con mayor capacidad para disminuir los niveles de cLDL.
Otro de los fármacos utilizados, es la ezetimiba, la cual inhibe la absorción de colesterol a nivel intestinal. Es un complemento y en ocasiones incluso sustitutivo de las estatinas.
La ezetimiba se usa junto con CTEV (dieta, adelgazamiento, ejercicio) para reducir la cantidad de colesterol y otras grasas presentes en la sangre.
Las resinas disminuyen el cLDL pero tienen el inconveniente de que pueden elevar las cifras de TGs y producir intolerancia digestiva, no obstante, en niños con HF, es el fármaco de elección.
Los fibratos en cambio, tienen menos efecto en disminuir las cifras de cLDL a pesar de descender los TGs y aumentar el HDLc.
Ensayos clínicos realizados en PP han demostrado eficacia en la disminución de enfermedad coronaria y mortalidad coronaria en población con riesgo coronario medio superior al 12,5% a los 10 años.
En pacientes con CT <200 mg/dl se debe realizar cribado cada 5 años, y en aquellos con cifras>200 mg/dl realizaremos perfil lipoproteico considerando factores de riesgo emergentes (Lp(a), Fibrinogeno, Homocistiena, etc) y valoraremos el riesgo coronario. Si este riesgo es inferior al 10% a 10 años se aconseja medidas higienico-dieteticas y controles reevaluando el riesgo cada 5 años. En aquellos con riesgo mayor al 10% a los 10 años, se realizan medidas higiénico – dietéticas durante 3-6 meses y reevaluación. Si se alcanzan los objetivos de control, control semestral y en caso contrario, se aconseja el uso de tratamiento farmacológico.
El uso combinado de estatinas y fibratos no está recomendado, ya que se ha asociado al aumento del riesgo de miopatías, especialmente, en pacientes con insuficiencia renal, siendo más común el uso de la combinación estatina+ezetimiba que tiene menos efectos secundarios al necesitar menos dosis y actuar por doble mecanismo (hepático-intestinal).
Hasta los años 2000 se publicaron 5 ensayos clínicos de PS comparativos con placebo que demostraron la disminución de la mortalidad coronaria.
Los pacientes en PS, deben ser cribados anualmente mediante la determinación plasmática de CT, HDL, LDL y TGs. El objetivo de control en estos pacientes, será un cLDL <100mg/dl.
El tratamiento de la hipercolesterolemia con medidas higiénico – dietéticas es común en PP y PS, en cuanto al tratamiento farmacológico, debemos escoger la estatina más adecuada en función del nivel de reducción del cLDL. Es importante tratar la causa subyacente del problema (Hipotiroidismo, ingesta de fármacos, enfermedad renal o hepática, etc.).
Cuando con el tratamiento farmacológico pautado no consigamos el control esperado, siempre y cuando el paciente haya cumplido correctamente este, se considerara el uso de combinaciones de fármacos, la más recomendable desde el punto de vista de eficacia y seguridad, es el ezetimiba y estatinas.
Bibliografía
Schaefer EJ, Tsunoda F, Diffenderfer M, Polisecki E, Thai N, Asztalos B. The Measurement of Lipids, Lipoproteins, Apolipoproteins, Fatty Acids, and Sterols, and Next Generation Sequencing for the Diagnosis and Treatment of Lipid Disorders. [Updated 2016 Mar 29]. In: De Groot LJ, Beck-Peccoz P, Chrousos G, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.
Mata P, Alonso R, Ruiz A, Gonzalez-Juanatey JR, Badimón L, Díaz-Díaz JL, et al. Diagnóstico y tratamiento de la hipercolesterolemia familiar en España: documento de consenso. Aten Primaria 2015;47(1):56-65.
Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European society of cardiology (ESC) and European Atherosclerosis society (EAS) developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis. 2016;253:281-344.
Wierzbicki AS, Humphries SE, Minhas R, on behalf of the Guideline Development Group. Familial hypercholesterolemia: Summary of NICE guidance. BMJ. 2008;337:a1095.
Avis HJ, Hutten BA, Gagné C, Langslet G, McCrindle BW, Wiegman A, et al. Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol. 2010;55(11):1121-6.
Braamskamp MJ, Langslet G, McCrindle BW, Cassiman D, Francis GA, Gagné C, et al. Efficacy and safety of rosuvastatin therapy in children and adolescents with familial hypercholesterolemia: Results from the CHARON study. J Clin Lipidol. 2015;9(6):741-50.
Langslet G, Breazna A, Drogari E. A 3-year study of atorvastatin in children and adolescents with heterozygous familial hypercholesterolemia. J Clin Lipidol. 2016;10(5):1153-62.
Miyagi Y, Harada-Shiba M, Ohta T. Effect of Statin Therapy in 4-Year-Old Dichorionic Diamniotic Twins with Familial Hypercholesterolemia Showing Multiple Xanthomas. J Atheroscler Thromb. 2016;23(1):112-7.
Araujo MB, Pacce MS. A 10-year experience using combined lipid-lowering pharmacotherapy in children and adolescents. J Pediatr Endocrinol Metab. 2016 Oct 8. pii:/j/jpem.ahead-of-print/jpem-2016-0117/jpem-2016- 0117.xml.
Besseling J, Hovingh GK, Huijgen R, Kastelein JJ, Hutten BA. Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J Am Coll Cardiol. 2016;68(3):252-60.
Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. 2015;132(22):2167-92.
Maron DJ, Hartigan PM, Neff DR, Weintraub WS, Boden WE; COURAGE Trial Investigators. Impact of Adding Ezetimibe to Statin to Achieve Low-Density Lipoprotein Cholesterol Goal (from the Clínicasl Outcomes Utilizing Revascularization and Aggressive Drug Evaluation [COURAGE] Trial). Am J Cardiol. 2013;111:1557-62.