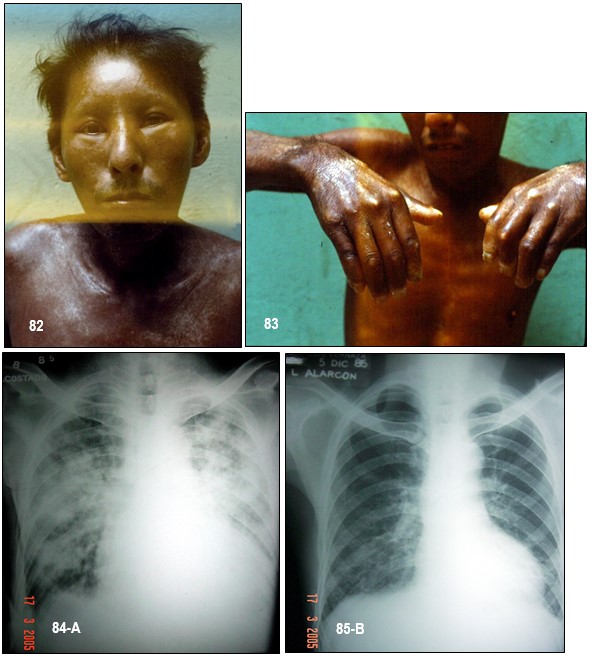
Imagen 82.- Esclerosis sistémica progresiva (ESP). Paciente de 55 años de edad que ingresa por presentar antecedentes desde hace más de 10 años de cambios en la tersura de su piel, asociado a hiperpigmentación en casi todo el cuerpo. Artralgias difusas. Refiere que toma esteroides desde hace 8 meses y que se hace 20 días aparece fiebre, decaimiento, y tos eventualmente con esputos hemoptoicos. Se realizaron estudios específicos incluyendo tuberculosis. La imagen muestra un paciente con la enfermedad activa, edema facial, y cambios esclerodermiformes e hiperpigmentación en toda la piel. Imagen 83.- ESP. Acroesclerosis. Nótese la severa esclerodactilia, o sea, la induración de la piel que recubre los dedos, tal como se ilustra en esta imagen. El paciente trata de extender sus dedos a lo máximo posible sin lograrlo. Es frecuente la aparición de úlceras en los pulpejos y sobre los nudillos que puede resultar invalidante debido a la atrofia cutánea y a la mutilación espontánea. Imágenes 84-85.- ESP. Tuberculosis en el curso de la corticoterapia. En A, múltiples lesiones infiltrativas densas y homogéneas dispersas en ambos pulmones indicativas de tuberculosis. En B, patrón radiológico al mes de haberse instaurado quimioterapia antifímica. Se evidencia mejoría radiológica que fue correlativa con la clínica del paciente. Es importante prever esta complicación durante la terapia con corticosteroides, con quimioprofilaxis con isoniazida. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
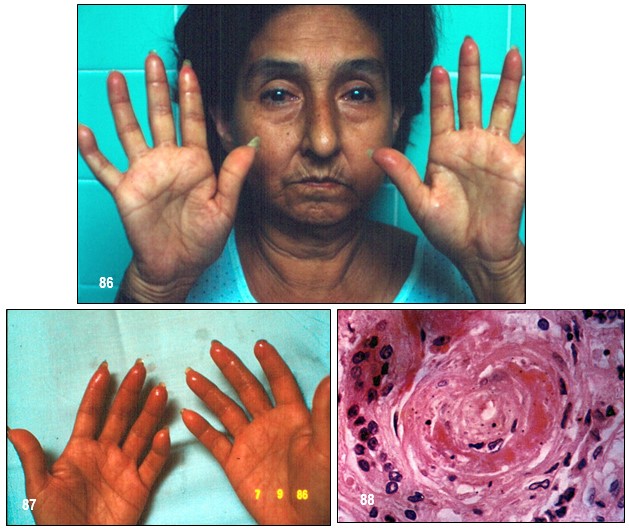
Imagen 86.- Esclerosis Sistémica Progresiva. Paciente de 62 años de edad con diagnóstico de esclerodermia desde hace aproximadamente 9 años. Ingresa por control de probable fracaso renal esclerodérmico (hipertensión arterial en estadio 2, movilización al alza de los nitrogenados). Se realizó biopsia renal. Obsérvese la fase vasodilatadora – hiperémica del fenómeno de Raynaud, con zonas de eritema perfectamente delimitadas en los pulpejos de todos los dedos. Pliegues radiados peribucales más acentuados en el labio superior “en bolsa de marinero”. Imagen 87.- Esclerosis Sistémica Progresiva. Fenómeno de Raynaud. El fenómeno de Raynaud es un evento de la microvasculatura de los dedos que suele preceder, aparecer conjuntamente o posterior a la esclerodermia. En este caso apareció a los 4 años de iniciarse las manifestaciones de básicas. En este fenómeno en un primer momento aparecen zonas blanqueadas perfectamente delimitadas, posteriormente hay cianosis y por último hiperemia reactiva; este fenómeno puede aparecer tanto en los pulpejos de los dedos como en la punta de la nariz e inclusive a nivel de los lóbulos de las orejas. Imagen 88.- Esclerosis Sistémica Progresiva. Vasculopatía de una crisis renal esclerodérmica. Biopsia renal. Arteria interlobulillar (flecha) que muestra un grave engrosamiento mucoide de la íntima con exudación de fibrina (depósitos amorfos rojos). El compromiso de la luz vascular determina un círculo vicioso de hipertensión maligna y empeoramiento de la microangiopatía trombótica (H & E.).
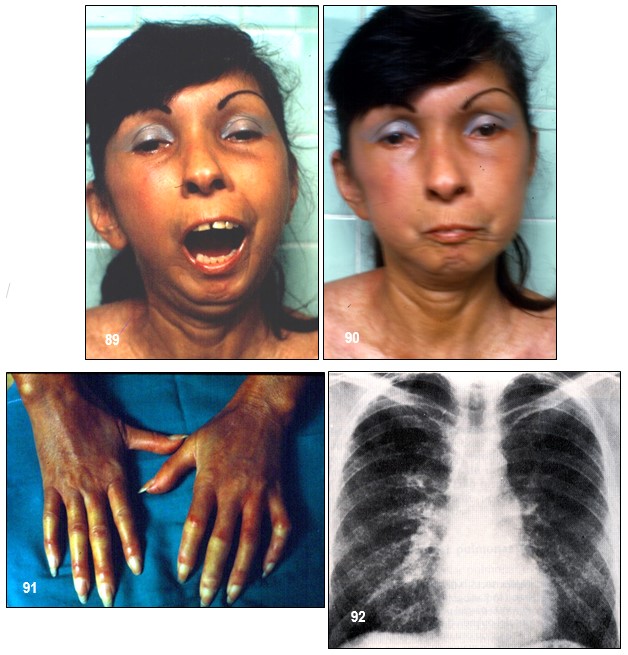
Imagen 89.- Esclerosis sistémica progresiva. Paciente de 44 años de edad diagnosticada de esclerodermia desde hace 7 años, bajo terapia sintomática. Ingresa para control dentro de un programa de evaluaciones periódicas. Por lo demás asintomática. En una placa de tórax se evidenció un patrón radiológico de fibrosis pulmonar. Imagen 90.- ESP. Facies característica, inexpresiva, “facies de pajarito”. Desaparición de las arrugas, afinamiento de la nariz y microstomía. Al pedirle que abra la boca presentaba limitación importante del movimiento por la apretada piel de la acroesclerosis. Nótese rágades peribucales propia de la atrofia cutánea (boca en bolsa de marinero). Imagen 91.- ESP. Acroesclerosis. Severa acroesclerosis con zonas de enrojecimiento en los dedos que contrasta con otras zonas de palidez marcada. Pequeñas úlceras a la altura de ciertos nudillos. Piel atrófica y brillante. Manchas hipo e hiperpigmentadas en «sal y pimienta» en la parte supero-anterior del tórax. Imagen 92.- ESP. Fibrosis pulmonar intersticial difusa. Patrón radiológico bilateral de trazado reticular de líneas finas que afecta especialmente las partes inferiores de ambos pulmones. Este tipo de lesión es frecuente encontrar en los casos avanzados de esta colagenosis, aquí se sugiere instaurar terapia esteroidea. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
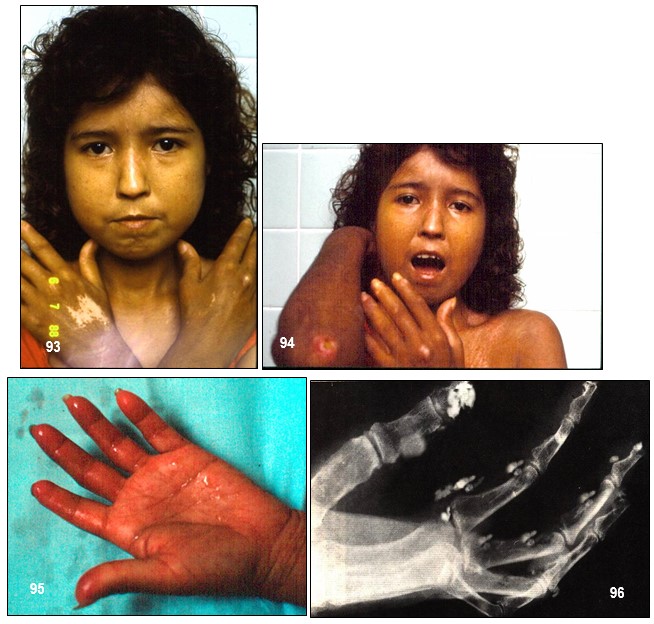
Imagen 93.- ESP. Paciente de 18 años de edad con antecedentes de esclerodermia desde hace 4 años. Nótese los rasgos faciales muy sugestivos de esta colagenosis. Además, máculas y manchas despigmentadas; acroesclerosis. Imagen 94.- ESP. Acroesclerosis. Cara tensa y brillante con labios apretados que imposibilita la total abertura de la boca. En segundo plano, se observan dos úlceras vasculíticas, una en el codo y otra en la articulación metacarpofalángica proximal, además, de manchas hipopigmentadas en el cuello y tórax anterior. Imagen 95.- ESP. Acroesclerosis. Engrosamiento de la piel de los dedos y de las palmas; se evidencia vasculitis acral. Imagen 96 .- ESP. Calcinosis de partes blandas. Depósitos de grupos amorfos de calcio en las partes blandas digitales. Nótese las alteraciones óseas tróficas alrededor de los extremos terminales. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
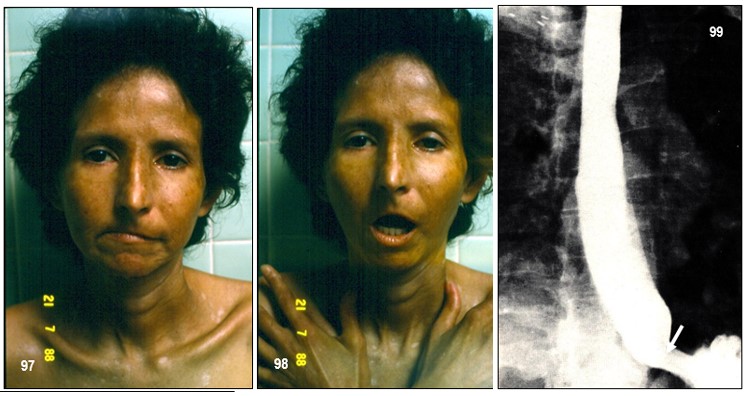
Imagen 97.- ESP. Paciente de 33 años de edad con antecedentes de esclerodermia desde hace 7 años. Nótese los rasgos faciales clásicos de esta colagenosis, muy similar a los casos anteriores. Obsérvese el rostro de sufrimiento que contrasta con manchas hipopigmentadas vitiligoides en el tórax superior y manos. Acusaba disfagia para sólidos y a veces para líquidos, a los que se agrega vómitos. Se solicitó estudio radiológico del esófago. Imagen 98.- ESP. Acroesclerosis. Imposibilidad para abrir la boca debido a la piel apretada y atrófica; algunas telangiectasias en los labios y acroesclerosis importante. Imagen 99.- ESP. Esfigmografía. Esófago dilatado y atónico. La unión gastroesofágica abierta (flecha), muy sugestivo de esófago esclerodérmico.
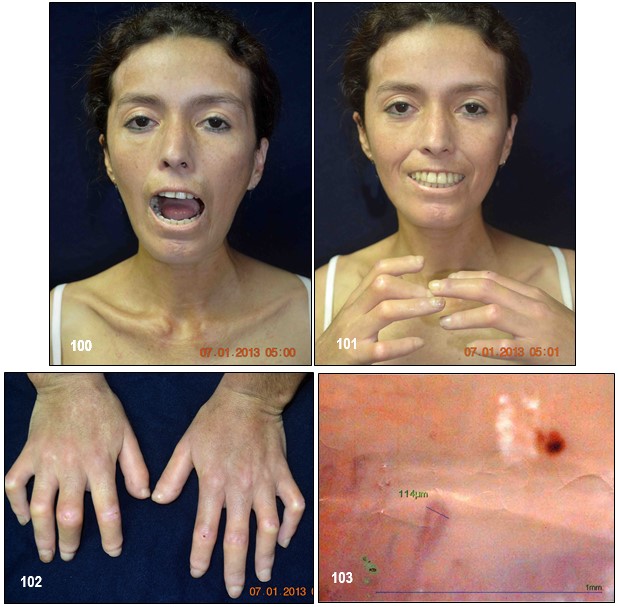
Imagen 100.- ESP. Paciente de 33 años de edad diagnosticada hace 7 años de ESP. Refería que su enfermedad debuta con edema y cambios de color de la piel de la cara, fatiga recurrente, malestar inespecífico, artromialgias (especialmente de antebrazos), febrículas, fenómeno de Raynaud de los dedos de las manos (en respuesta a exposición al frio), perdida ponderal e hiporexia, posteriormente en su evolución se agregaron otros síntomas como cambios en la textura de la piel, cuadros recurrentes de tos no productiva y cefaleas eventuales. Medicada de manera irregular con esteroides sistémicos, vasodilatadores periféricos, metotrexato, ácido acetilsalicílico, y otros sintomáticos. Adviértase dificultad y limitación para abrir completamente la boca, que contrasta con la esclerosis de la piel. Imagen 101.- ESP. Facies inexpresiva y cambios esclerodermiformes a nivel de ambas manos. Se realizaron exámenes. Imagen 102.- ESP. Acroesclerosis. Severa acroesclerosis con zonas de enrojecimiento en los nudillos de los dedos que contrasta con pequeñas ulceras de aspecto vasculíticas y reabsorción de las falanges de ambos dedos pulgares. Piel atrófica y brillante. La extensa fibrosis subcutánea prácticamente ha inmovilizado los dedos de las manos, creando una deformidad en flexión similar a una garra. La pérdida de vascularización ha dado lugar a ulceraciones cutáneas. Imagen 103.- ESP. Capilaroscopia. Por videocapiloroscopia a nivel del pliegue periungueal de ambas manos, el número de capilares se encuentra disminuido, capilares dilatados que conforman megacapilares, zonas vasculares y hemorragias abundantes. Nota: La pérdida progresiva de capilares ha sido asociada a compromiso cutáneo extenso, ulceraciones cutáneas y peor pronóstico en pacientes con esclerodermia sistémica. Véase el estudio radiológico y tomográfico de pulmones en las imágenes 104, 105,106 y 107. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
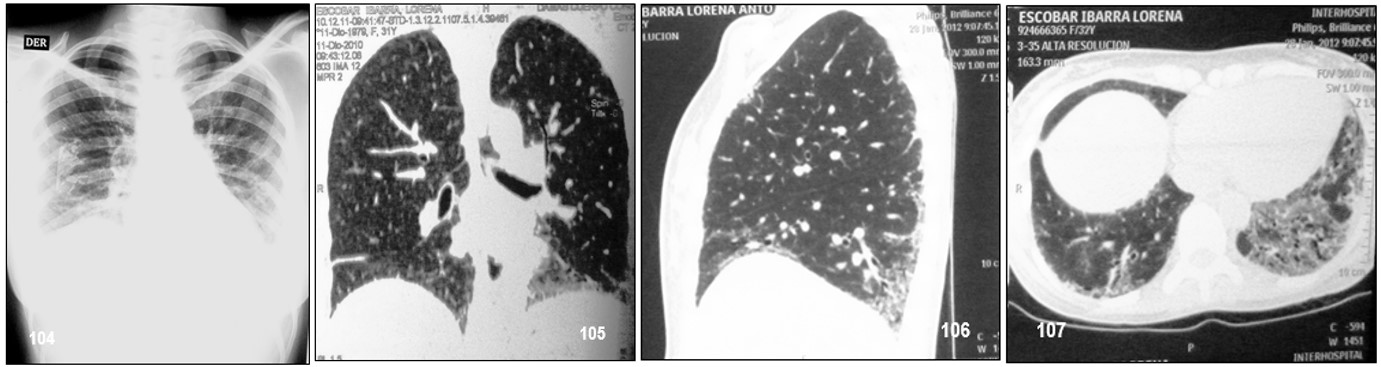
Imagen 104.- ESP. Radiografía AP de tórax: Infiltrados intersticiales en “panal de abejas” más evidentes en las bases pulmonares. Imagen 105.- ESP. TAC simple de pulmones: Se realizó TAC de tórax multiplanar desde los vértices pulmonares hasta las cúpulas diafragmáticas con reconstrucción bidimensional, coronal y sagital y cortes de 0,6 mm. Infiltrados intersticiales productivos exudativos más patrón alveolar y dilatación de bronquiolos de las bases pulmonares. Trama pulmonar bilateral acentuada. Tráquea y bronquio fuertes principales de aspecto tomográfico normal. Corazón y grandes vasos de diámetros normales. No hay crecimiento ganglionar de la cadena para-aortica. Imagen 106 .- ESP.TAC: Imágenes desde el opérculo torácico hasta las cúpulas diafragmáticas con protocolo de alta resolución. Áreas de panalización asociada a bronquiectasias por tracción, así como imágenes cilíndricas y varicoides, predominando hacia los segmentos basales de ambos pulmones con engrosamiento del tabiques interlobulillares asociadas a imágenes de vidrio deslustrado con engrosamiento pleurales focales regionales y engrosamiento septales periféricos, subpleurales. Imagen 107.- ESP, TAC de alta resolución: Manifestaciones descritas en la imagen 106, más esófago dilatado. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
INFORMACIÓN BÁSICA: ENFERMEDADES VASCULARES DEL COLÁGENO: Esclerosis Sistémica Progresiva (ESP) (ORPHA:220393). Variante CREST (ORPHA:90290).- Es un enfermedad del tejido conjuntivo que involucra cambios en la piel, los vasos sanguíneos, los músculos y los órganos internos. Es un tipo de trastorno autoinmunitario, una afección que ocurre cuando el sistema inmunitario ataca por error y destruye tejido corporal sano. Se desconoce la causa de la ESP. Los pacientes con esta enfermedad presentan una acumulación de una sustancia llamada colágeno en la piel y otros órganos. Esta acumulación lleva a que se presenten síntomas de la enfermedad. La enfermedad generalmente afecta a personas de 30 a 50 años de edad y las mujeres la desarrollan más a menudo que los hombres. Algunas pacientes con ESP tienen antecedentes de estar alrededor del polvo de sílice y policloruro de vinilo, pero la mayoría no. La ESP, puede ocurrir con otras enfermedades autoinmunitarias, entre ellas lupus eritematoso sistémico y polimiositis. En tales casos, el trastorno se denomina enfermedad mixta del tejido conjuntivo. Algunos tipos de esclerodermia afectan sólo la piel, mientras que otros afectan todo el cuerpo.
La ESP es una enfermedad generalizada del tejido conectivo que se caracteriza por engrosamiento y fibrosis de la piel y que compromete a órganos internos como el tracto gastrointestinal, pulmón, corazón y riñón. Se acompaña de anticuerpos relativamente específicos y de alteraciones de la microvasculatura. Su morbilidad y mortalidad son considerables y se relacionan con el compromiso visceral. La enfermedad es heterogénea en su extensión, progresión y en el compromiso de órganos internos.
Epidemiología.- No es una enfermedad muy frecuente. Sin embargo la exposición a agentes ambientales puede desencadenar enfermedades del tipo de la esclerodermia. Algunos de estos desencadenantes ambientales son: exposición a sílice; a vibraciones (bull-dozers); a solventes como el cloruro de vinilo o solventes orgánicos; a silicona mediante implantes mamarios; a enfermedad injerto contra huésped del transplante de médula ósea. Recientemente hubo dos epidemias de cuadros Es símil; una ocurrió en España en 20.000 personas que ingirieron aceite de raps adulterado y la otra fue en EEUU en 1.500 personas que ingirieron tabletas contaminadas de L-triptofano y que desarrollaron un síndrome de Eosinofilia-Mialgia.
Patología.- Lo característico es fibrosis de la piel, de los vasos y de otros órganos.
Piel.- En la dermis hay aumento de colágenos y de otros constituyentes de la matriz, se adelgaza la epidermis y desaparecen los clavos córneos. En la fase inicial de las lesiones cutáneas se observan infiltrados de células, linfocitos y monocitos. En la fase tardía hay atrofia de la piel, la que es relativamente acelular.
Vasos.- Las lesiones vasculares se observan en todos los órganos afectados por la ES. La enfermedad de la microcirculación es diseminada y se puede observar in vivo en el enfermo al examinar con buena luz y una lupa de aumento, a veces a simple vista, el lecho capilar del pliegue de la piel que bordea las uñas. Se ven en la cutícula puntos rojos que corresponden a capilares dilatados o a hemorragias capilares y áreas avasculares. Los vasos grandes muestran hiperplasia de la íntima que puede llegar a obliterar el lumen.
Patogénesis.- En la forma generalizada de la ES hay una alteración difusa de la microcirculación que da como resultado una fibrosis intensa y no regulada. El sello de la enfermedad es un proceso de cicatrización de heridas no controlado.
Lesiones arteriolares y capilares preceden a la fibrosis. El pronóstico del paciente depende principalmente de la intensidad y de la rapidez de aparición de estas lesiones en órganos vitales como riñones, pulmones, corazón e intestino.
Fibrosis.- Depósito ininterrumpido de matriz extracelular en la íntima de los vasos sanguíneos, en el espacio pericapilar y en el intersticio de la piel por fibroblastos activados. Una de las moléculas solubles involucradas en gatillar este estado de activación de fibroblastos es el factor de crecimiento TGF-b (transforming growth factor beta).
Inmunidad.- Aunque no se conoce el papel de los autoanticuerpos en la patogénesis de la ES, el 80-90% de los pacientes con ES presenta algún autoanticuerpo. Son específicos para esclerodermia el anticuerpo anticentrómero (antígeno blanco en el núcleo, en el kinetocoro; es marcador de la variedad limitada o CREST) y el antitopoisomerasa I (antígeno es la enzima DNA.topoisomerasa; marcador de la variedad difusa). Además de esos anticuerpos antinucleares, se encuentran anticuerpos dirigidos contra estructuras de la matriz como anticuerpos anti tipos de colágeno IV y V.
Se ha identificado una respuesta exagerada de los linfocitos T CD4 (ayuda) a citoquinas (IL-2) lo que sería crucial en la activación de fibroblastos. De acuerdo con esas observaciones, las extensas lesiones vasculares de la esclerodermia que se caracterizan por activación del endotelio, daño y proliferación de la íntima pudieran ser explicadas por:
- Una lesión inicial por agresión citolítica de las células T a células endoteliales;
- La continuación del daño por autoinmunidad contra antígenos de la estructura vascular, como el colágeno tipo IV y la laminina.
Clasificación.- La Esclerodermia y los Síndromes esclerodermia símil se clasifican en la tabla 9.

Manifestaciones clínicas.- Esclerosis sistémica. Existen 2 formas clínicas cuya presentación inicial puede ser similar
ESP Difusa: Progresión rápida de fibrosis cutánea. Da compromiso proximal de extremidades, tronco y cara y compromiso visceral precoz. Es grave y poco frecuente. Tiene como autoanticuerpo marcador a antitopoisomerasa I (Scl-70). El pronóstico general es malo.
ESP Limitada o CREST: Progresión lenta. Fibrosis limitada a distal (manos, pies) y a cara. El compromiso visceral es tardío. Tiene como autoanticuerpo marcador a anticentrómero. El pronóstico es mejor. El fenómeno de Raynaud es el primer síntoma en el 95% de los enfermos, precede a veces por muchos años la aparición de ES. Varios meses antes que sea claramente distinguible el engrosamiento de la piel aparecen edema de dedos y manos o artralgias o artritis de las pequeñas articulaciones de las manos en frecuencia variable.
Afectación vascular: La afectación de las arterias digitales origina el denominado fenómeno de Raynaud, que consiste en ataques episódicos vasoespásticos, con palidez y/o cianosis (coloración amoratada) de los dedos seguidos de hiperemia por reperfusión. En una secuencia de coloración blanca, amoratada y roja, muy característica. Los ataques suelen desencadenarse tras la exposición al frío o a tensión emocional. Suele existir afectación bilateral de los dedos de la mano y también del pie. Aparece en el 100% de las formas de esclerodermia limitada y hasta en el 75% de las formas difusas. Pueden llegar a originar infartos en los pulpejos de los dedos con ulceraciones o gangrena evidente, con riesgo de complicaciones de tipo infeccioso, que pueden incluso afectar al hueso subyacente. Su ausencia se ha descrito asociada a un mayor riesgo de afectación renal. No sólo ha de atribuirse a fenómenos vasoespásticos, sino también a la lesión estructural de los vasos sanguíneos.
Dicha lesión estructural es visible mediante una técnica, la denominada capilaroscopia periungueal, cuya importancia radica en que en los pacientes con ESP pueden verse dos patrones de alteración capilar que pueden predecir en meses e incluso en años la forma clínica antes de que aparezca otra sintomatología clínica.
Dichos patrones son:
- a) patrón lento, con dilatación de asas capilares por sus tres porciones arteriolar, capilar y venular, y
- b) patrón activo, con pérdida de asas capilares por destrucción de las mismas ya sea difusamente o de forma más típica en áreas vecinas a las zonas de dilatación.

Imagen 11 con fines Didácticos: Severa esclerodactilia que ha deformado los dedos con pérdida espontánea de la falange distal del dedo índice de la mano derecha. Varios de ellos en riesgo de evolucionar a la gangrena. La paciente presentaba episodios vasculares de Raynaud. Paciente con ESP variante CREST. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
Video que muestra los cambios de color de la piel al exponerse a agua fria.
Un paciente que presenta únicamente fenómeno de Raynaud con un patrón lento en la capilaroscopia tiene un riesgo elevado de padecer una Esclerosis Sistémica Limitada sobre todo si se evidencia positividad para anticuerpos anticentrómero. Institut Ferran de Reumatologia, dispone de avanzados equipos de capilaroscopia y profesionales entrenados en la detección y clasificación de estas anomalías.
Existen otras técnicas que permiten comprobar el grado de afectación vascular en los pacientes con Esclerosis Sistémica. Estas técnicas son útiles pero no esenciales. Nos referimos a los estudios doppler color (una avanzada técnica mediante ultrasonidos) para medir el flujo de arterias radiales, palmares y digitales; a pruebas de provocación con frío; y a la determinación de los niveles plasmáticos de determinadas moléculas (moléculas solubles de adhesión como ICAM-1 o VCAM-1, endotelina-1, óxido nítrico, prostaciclina o moléculas del sistema de coagulación y fibrinólisis).
Pero para la evaluación del grado de afectación vascular, el método más fiable es determinar una serie de parámetros que tienen que ver con el fenómeno de Raynaud y sus consecuencias. Respecto al fenómeno de Raynaud, se debe determinar su duración, frecuencia, momento de aparición y gravedad. Con relación a las úlceras isquémicas, se evaluará su presencia o ausencia, tamaño, actividad y número de dedos afectos.
Las alteraciones en la microcirculación son también responsables de una manifestación cutánea típica de la Esclerosis Sistémica como son las telangiectasias que acostumbran a aparecer en dedos, palmas de las manos y cara, y típicamente en pacientes con la forma limitada. Además de en la piel pueden localizarse en la mucosa oral y del tracto gastrointestinal. Las alteraciones vasculares pueden afectar, aunque de forma mucho más inusual, a vasos de gran calibre habiendo sido implicadas en la etiopatogenia de la hipertensión pulmonar no asociada a afectación del parénquima pulmonar, la crisis renal esclerodérmica y el denominado Raynaud coronario (una forma rara de presentación que recuerda la angina de pecho).
– Afectación cutánea: Aparece prácticamente en el 100% de los pacientes (aunque, como he comentado antes, existe una entidad denominada esclerosis sistémica sine scleroderma en la que hay afectación orgánica sin lesión cutánea).
El endurecimiento de la piel en las zonas proximales a las articulaciones metacarpofalángicas o acroesclerosis proporciona el diagnóstico definitivo en el 90% de los casos; sin embargo, la esclerodactilia por sí sola no permite el diagnóstico, ya que puede estar relacionada con otros procesos como la enfermedad de Raynaud u otras alteraciones de la circulación periférica.
En los pacientes con formas limitadas, la afectación cutánea comienza por los dedos de las manos y pies, y evoluciona lentamente durante años a antebrazos, cara y cuello. En los pacientes con formas difusas progresa rápidamente durante meses, desde manos y pies a antebrazos, piernas, muslos y tronco. La lesión cutánea evoluciona típicamente en tres fases: la edematosa, esclerótica o indurativa y la atrófica. En la fase edematosa, la piel está tensa con edema difuso e indoloro y pueden aparecer cambios de coloración.
En la fase indurada el edema es reemplazado por un engrosamiento duro de la dermis que hace que la piel se adhiera a las estructuras profundas y pierda los pliegues cutáneos, dificultando la movilidad normal, especialmente la extensión de los dedos y la apertura bucal; la epidermis se adelgaza, por lo que la piel afecta es generalmente fina y brillante. Los anejos cutáneos desaparecen perdiéndose pelo y desapareciendo la sudoración en las extremidades. Pueden aparecer trastornos de la pigmentación con hiperpigmentación, acromia o la combinación de ambas, por lo que la piel pude adoptar un aspecto moteado. Los dedos se afilan progresivamente y pueden aparecer úlceras en zonas acras o sobre prominencias óseas dado que son zonas más vulnerables a traumatismos, que son úlceras de difícil tratamiento debido a la piel donde asientan y a su escasa vascularización. En la fase atrófica, el engrosamiento dérmico desaparece y vuelven a aparecer pliegues cutáneos aunque la epidermis y los anejos cutáneos permanecen atróficos.

Imágenes 12 con fines Didácticos: Imagen izquierda: Histopatología de piel: Vista panorámica: La afectación de la forma en placa está centrada en dermis reticular baja mientras que otras variantes presentan reemplazo de la grasa y tejidos subyacentes por colágeno. La epidermis suele ser normal. En los primeros estadios inflamatorios se observa en dermis reticular y /o estructuras subcutáneas infiltrados intersticiales de linfocitos y ocasionalmente eosinófilos. Las paredes de de los vasos muestran edema endotelial y engrosamiento haces colágenos preexistentes y depósito de fibras de colágeno recientemente formado. Imagen derecha: Severa acroesclerosis con zonas de enrojecimiento en los dedos que contrasta con otras zonas de palidez marcada. Pequeñas úlceras a la altura de algunos nudillos. Piel atrófica y brillante. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
Cuando la enfermedad se inicia en los antebrazos, brazos o tórax, la piel se torna brillante, nacarada y ofrece cierta resistencia a ser plegada. El cuello puede afectarse de forma precoz y mostrar pliegues endurecidos a modo de bandas, cuando se fuerza la extensión del mismo (es el conocido como «signo del cuello»).
La cara adquiere también un aspecto muy característico, con pérdida de los pliegues fisiológicos con un aspecto de seudorrejuvenecimiento en estadios iniciales. Posteriormente se pierden las líneas de expresión, la nariz se torna pequeña y afilada, los párpados inferiores se atrofian y endurecen, lo que puede dificultar el cierre completo de los ojos. Se reduce la apertura bucal (microstomía) y, aparecen pliegues radiales peribucales. En las mejillas, dorso de nariz, frente y labios pueden observarse las típicas telangiectasias.
La calcinosis consiste en el depósito de calcio a nivel subcutáneo, asentando sobre prominencias óseas de dedos, antebrazos, alrededor de codos y rodillas en cresta ilíaca y zonas paravertebrales. La piel donde asientan se torna eritematosa y acaba ulcerándose, dando salida a un material blanco y espeso constituido por cristales de hidroxiapatita. La causa de la calcinosis es desconocida y no se han registrado alteraciones en el metabolismo fosfocálcico.
En contraste en el CREST, o forma limitada, la fibrosis se limita a los dedos, o a los dedos y a la cara. No presenta compromiso de piel sólo un 2% de los enfermos (esclerodermia sine esclerodermia). Aparecen telangiectasias y calcinosis.
– Afectación gastrointestinal: Tiene lugar en la mayoría de los pacientes y difiere poco en los dos subgrupos de ESP. Puede adoptar muchas formas clínicas que se pueden presentar aisladas o asociadas entre ellas.
La afectación esofágica se acerca al 80% de los casos, siendo más frecuente la disfunción motora del tercio distal. La debilidad e incoordinación de la musculatura lisa esofágica generan disfagia (dificultad al tragar) especialmente a sólidos. Así mismo, la afectación del esfínter esofágico inferior condiciona la aparición de reflujo gastroesofágico con esofagitis péptica, ulceraciones y estenosis. Los cambios histológicos incluyen atrofia y sustitución fibrosa de la muscularis y depósito de colágeno en la lámina propia y la submucosa.
Otras complicaciones esofágicas que se observan en estos pacientes son la sobreinfección por el virus herpes simple, citomegalovirus y Cándidas, sobre todo en los paciente ya inmunocomprometidos.
El tránsito gastrointestinal puede mostrar no solo la presencia de trastornos en la motilidad sino también la existencia de reflujo y sus potenciales complicaciones. Pero hay muchos pacientes asintomáticos o que solo refieren disfagia, en los cuales la práctica de un tránsito esofagogastroduodenal no es de gran utilidad. En dichos casos la práctica de una manometría esofágica (con monitorización del pH a nivel distal de esófago) puede ayudar a predecir los pacientes con más riesgo de presentar reflujo gastroesofágico, por lo que actualmente se considera la prueba más importante para documentar la magnitud de la afectación esofágica. La endoscopia esofágica demostrará la existencia de complicaciones asociadas al reflujo, como la esofagitis péptica, los fenómenos de metaplasia o la esofagitis infecciosa. Ante todo paciente afecto de ESP sin síntomas o con disfagia sin clínica de reflujo o cuando éste exista pero no se detecte en el TEGD, está indicada la práctica de una manometría esofágica con monitorización del pH esofágico.
La afectación del estómago en la ESP es poco frecuente, aunque pueden existir trastornos en la motilidad y sobre todo la aparición de telangiectasias (que pueden localizarse a cualquier nivel de la mucosa digestiva) con pérdidas crónicas que originen anemia o episodios agudos de hemorragia digestiva alta.
La aparición de aclorhidria secundaria (falta de ácido) a terapia antisecretora agresiva para el tratamiento del reflujo gastroesofágico, puede producir un incremento de bacterias gástricas y cambios en la flora habitual del intestino con alteraciones del ritmo deposicional.
El duodeno se afecta con mayor frecuencia que el estómago, dando lugar a sensación de plenitud postprandial, náuseas, vómitos y anorexia. De forma excepcional, la fibrosis duodenal extensa puede dar lugar a malabsorción de hierro.
La hipomotilidad del yeyuno e íleon se debe a pérdida de músculo liso pero es poco frecuente pues afecta a un 20% de los pacientes. El sobrecrecimiento bacteriano es una complicación frecuente de la atrofia de la musculatura lisa que da lugar a malabsorción y posterior malnutrición. Clínicamente se manifiesta en forma de episodios intermitentes de distensión abdominal marcada, diarrea, gran pérdida de peso y episodios dolorosos de seudoobstrucción intestinal. Existe malabsorción de grasas (probablemente secundaria a un déficit de conjugación por falta de sales biliares), de hidratos de carbono y de vitamina B12; todo ello objetivable a través de las pruebas de la D-xilosa, trioleína marcada y de Shilling. Analíticamente se detectan marcadores bioquímicos de malabsorción como hipoalbuminemia e hipocalcemia.
Las alteraciones en el colon pueden observarse en un 10%-50% de pacientes afectos de esclerodermia, constituyendo el enema opaca una prueba diagnóstica fundamental. El estreñimiento es secundario a hipomotilidad colónica y algunos pacientes presentan incontinencia o prolapso rectal secundario a incompetencia del esfínter rectal. Se puede producir impactación fecal o de contraste baritado en los divertículos con aparición de ulceraciones y el potencial riesgo de perforación. También puede aparecer dilatación colónica, pero el desarrollo de megacolon es raro.
– Afectación renal: Aunque la crisis renal aguda y grave acontece en un 10% de los pacientes, hasta un 50% presentan algún tipo de disfunción renal como proteinuria o hematuria microscópica, alteraciones en el sedimento, hipertensión arterial o disminución del filtrado glomerular (clearance de creatinina), con azotemia. En series necrópsicas hasta un 80% de los pacientes presentan lesiones histológicas vasculares a nivel renal.
La crisis renal esclerodérmica era, antes de la aparición de los fármacos IECA (Inhibidores del Enzima Convertidor de la Angiotensina), la principal causa de muerte en estos pacientes. Aparece en pacientes con Esclerosis Sistémica Difusa generalmente al inicio (menos de 4 años desde la aparición de los síntomas) y suele hacerlo en fases de afectación de induración dérmica rápidamente progresiva.
A veces comienza bruscamente hipertensión maligna, hiperreninemia y falla renal oligúrica. Hasta antes que aparecieran los inhibidores de la enzima convertidora de la angiotensina, la enfermedad renal con crisis hipertensiva era la causa de muerte en 1/3 de los pacientes.
Es poco frecuente en pacientes con la forma limitada. Sin una fase previa, el paciente desarrolla una hipertensión arterial maligna (aunque en ocasiones las cifras de tensión arterial pueden ser normales lo que con frecuencia se asocia a anemia hemolítica microangiopática grave con trombocitopenia) y fallo renal oligúrico agudo.
Habitualmente las crisis renales esclerodérmicas aparecen en épocas frías, por lo que se ha sugerido la existencia de un fenómeno de Raynaud renal. Otros factores capaces de desencadenar crisis renales son los corticoides y las depleciones rápidas de volúmenes sobre todo en relación con la utilización de diuréticos.
Se ha descrito una asociación entre cirrosis biliar primaria y CREST
– Afectación pulmonar: La afectación intersticial y la hipertensión pulmonar no asociada a alteración parenquimatosa son las de más repercusión clínica y pronóstica.
La afectación intersticial es la variedad más común, presentándose según diferentes series necrópsicas en más del 70% de los casos. Aparece con más frecuencia en pacientes con ESP y fumadores, así como en la forma CREST, y se asocia con síndrome de Raynaud grave, úlceras digitales y roces de fricción tendinosos. Clínicamente cursa con disnea de intensidad variable que puede llegar a ser de reposo junto con tos seca irritativa. El curso acostumbra a ser lento e insidioso, aunque en algunas ocasiones puede ser rápidamente progresivo.
En muchas ocasiones la radiografía simple de tórax puede ser normal. La tomografía axial computarizada (TAC) de tórax de alta resolución demuestra que hasta un 44% de pacientes con radiografía normal pueden mostrar alteraciones en la TAC con la consiguiente repercusión desde el punto de vista terapéutico. Además, esta prueba nos permite diferenciar las áreas de alveolitis de las de fibrosis establecidas. La alteración funcional más sensible y precoz es, de todas formas, la disminución en la capacidad de difusión (DLCO) que en ocasiones existe incluso antes de que se manifieste una alteración ventilatoria o se demuestre fibrosis por las pruebas de imagen.
Con el tiempo el funcionalismo pulmonar demuestra la existencia de una alteración ventilatoria de tipo restrictivo y/o obstructivo.
Con el tiempo estos pacientes desarrollan hipertensión pulmonar secundaria. En el lavado broncoalveolar puede haber un aumento de linfocitos o neutrófilos.
La hipertensión pulmonar no asociada a afectación parenquimatosa aparece en el 10% de los pacientes con la forma limitada y de muchos años de evolución. Clínicamente se caracteriza por disnea intensa y rápidamente progresiva en ausencia de afectación parenquimatosa. Se observa una reducción marcada de la DLCO (por debajo del 50% de lo normal previsible). Se puede detectar precozmente mediante ecocardiografía doppler, ya que el grado de insuficiencia tricuspídea se correlaciona linealmente con el pico de presión de la arteria pulmonar; por esta razón la práctica de esta técnica se tendría que considerar en todos los pacientes con Esclerosis Sistémica Limitada de muchos años de evolución, dado el pronóstico vital que el diagnóstico de esta entidad conlleva. Sin embargo, la prueba diagnóstica que nos permite no solo determinar la existencia de hipertensión arterial pulmonar y su grado, sino también la reactividad a determinados fármacos de los vasos pulmonares, es el estudio de hemodinámica pulmonar.
– Afectación cardiaca: La fibrosis miocárdica es frecuentemente asintomática y el SPECT de perfusión con talio es la prueba más sensible para detectarla. Puede, sin embargo, llegar a producir insuficiencia cardíaca congestiva de difícil tratamiento y control, además de arritmias ventriculares, con una mortalidad elevada.
La afectación fibrótica del tejido de conducción puede llegar a producir arritmias y bloqueos detectables hasta en un 50% de los pacientes con ESP. Estas alteraciones se manifiestan clínicamente en los pacientes afectos de ESP en forma de palpitaciones, lipotimias, presíncopes o síncopes.
Se han descrito infartos de miocardio en pacientes con ESP y coronarias morfológicamente normales. Los datos de los que se dispone actualmente sugieren que la combinación de factores que incluyen vasoespasmo coronario, fibrosis miocárdica, obliteración de las pequeñas arterias y de los capilares sería la responsable de la isquemia y necrosis.
Si el fenómeno vasoespástico contribuye de forma significativa, las repercusiones terapéuticas son evidentes, dado que los fármacos vasodilatadores son un tratamiento esencial en la ESP. La pericarditis es frecuente (35%-75% de las series necróspsicas y ecográficas) pero clínicamente sólo es evidente en un 10%-15% de los pacientes. La pericarditis crónica suele cursar clínicamente en forma de edemas persistentes en zonas declives.
En conclusión, la afectación cardíaca en la ESP es más frecuente en la forma difusa y puede manifestarse a nivel miocárdico, pericárdico, del sistema de conducción o en forma de arritmias. La aparición de manifestaciones clínicas (en aproximadamente un 10% de los pacientes) es un factor de mal pronóstico. Las alteraciones cardíacas asintomáticas son muy frecuentes. La afectación vascular no está aclarada, a diferencia de lo que ocurre en otros órganos. Aparte de la clínica, la evaluación de la afectación cardíaca en los pacientes incluye la práctica de un electrocardiograma (ECG), radiografía de tórax, Holter-ECG, y ecocardiografía doppler. El tratamiento es fundamentalmente sintomático y empírico.
– Afectación musculoesquelética: La contractura articular secundaria al engrosamiento y retracción dérmicos es la forma de afectación articular más frecuente en la Esclerosis Sistémica.
Pueden aparecer poliartralgias (dolores articulares difusos) que afectan tanto a grandes como a pequeñas articulaciones y son especialmente frecuentes al inicio de la enfermedad, para estabilizarse posteriormente. Sin embargo, la artritis es infrecuente, aunque ocasionalmente puede ser muy intensa y erosiva.
La afectación tendosinovial se manifiesta en forma de síndrome del túnel carpiano y aparece a la palpación de forma característica, como roces de fricción de los tendones que trasmiten una sensación correosa, predominantemente sobre muñecas, dedos, rodillas o tobillos; aparece en el 60% de los pacientes con Esclerosis Sistémica Difusa y en menos del 10% de los pacientes con Esclerosis Sistémica Limitada, lo que constituye un signo de gran valor diagnóstico.
Radiológicamente pueden observarse alteraciones óseas como reabsorción de falanges distales debida a osteólisis por hipovascularización-isquemia, semejando una artritis mutilante y que también puede afectar a la parte distal del radio y cúbito, la mandíbula y la porción superior de las costillas posteriores; osteoporosis yuxtaarticular y osteopoiquilia (véase Imagen con fines Didácticos).
La afectación muscular más frecuente es la atrofia por desuso secundaria a la limitación en la movilidad articular generada por la afectación cutánea, articular o tendinosa. Sin embargo, existen dos formas de miopatía clara. La primera de ellas consiste en una afectación muscular proximal no progresiva con elevación mínima de la creatin-fosfocinasa (CPK) secundaria a una sustitución de miofibrillas por tejido fibrótico sin signos inflamatorios en la biopsia muscular. Algunos pacientes, sin embargo pueden llegar a desarrollar una miopatía inflamatoria clásica indistinguible de una poli/dermatomiositis que suele aparecer en fases iniciales de la enfermedad, sobre todo en la forma difusa.
– Fibrosis.- El depósito de MEC en exceso es causante en gran medida de la morbimortalidad de la ES. Los fibroblastos y los miofibroblastos activados depositan tejido conjuntivo fibroso, de modo que la fibrosis reemplaza gradualmente a la fase inflamatoria, modifica la arquitectura del tejido dañado y produce la mayoría de los síntomas.
En la piel, la fibrosis comienza en la dermis profunda y en la parte superficial del tejido celular subcutáneo (TCS), y aumenta a medida que desaparece la microvasculatura y se destruyen los anejos. Desde los estadios tempranos de la enfermedad parecen establecerse pequeñas subpoblaciones autónomas de fibroblastos que producen un exceso de MEC. Se ha observado que estas subpoblaciones se suelen hallar cerca de células inflamatorias mononucleares o adyacentes a vasos sanguíneos. Son iniciadores de este proceso numerosas citoquinas y factores de crecimiento de múltiples vías de señalización.
Los pericitos y las células musculares lisas son células presentes en los pequeños vasos. Los pericitos tienen la capacidad de transformarse en células musculares lisas de la pared vascular, fibroblastos y miofibroblastos, y participar en la proliferación de células endoteliales. El aumento del grosor de la pared vascular, producido por la proliferación de las células musculares lisas vasculares, indica que las células están respondiendo al daño inducido por la ES. Los pericitos de las lesiones sobreexpresan numerosos receptores de citoquinas, incluyendo el receptor del PDGF. Las células endoteliales son las únicas células mesenquimales que entran en apoptosis en la ES temprana, mientras que las células musculares lisas vasculares y los pericitos proliferan profusamente.
Los fibroblastos son los encargados de la producción, el depósito y el remodelamiento de las fibras colágenas y de otros componentes de la MEC. Los fibroblastos en la ES se pueden convertir en miofibroblastos, y sobreexpresan diversas citoquinas (factor de crecimiento transformante β [TGF-β]) y receptores del TGF-β. Contienen también en la ES un exceso de especies reactivas de oxígeno (ROS). Estos fibroblastos tienen un defecto inherente en las microfibrillas que contienen fibrilina-1 y presentan sobreexpresión autónoma y mantenida del gen del colágeno tipo i.
TGF-β. La isoforma 1 desempeña un importante papel en la fibrosis, ya que es un mitógeno indirecto de los fibroblastos, mientras que las isoformas 2 y 3 parecen tener efectos antifibróticos. También es el más potente inductor de miofibroblastos y modula la expresión de varios receptores de citoquinas, incluyendo el propio TGF-β y el PDGF. Es causante de la elevada expresión del factor de crecimiento de tejido conectivo (CTGF), con actividad biológica similar al TGF-β. Una expresión aumentada de TGF-β y CTGF se ha detectado en lesiones iniciales de esclerodermia, pero después disminuyen los niveles del TGF-β y se mantienen elevados los del CTGF.
Inicialmente, el TGF-β se produce en células mononucleares del infiltrado inflamatorio, pero posteriormente lo producen los fibroblastos mediante regulación autocrina, en respuesta al TGF-β mediante la expresión de más receptores para el PDGF-α y el TGF-β1 en su superficie. Un circuito autocrino del TGF-β podría explicar la activación persistente de la expresión de genes de colágeno en fibroblastos, y sería causante de la naturaleza progresiva de la enfermedad.
Aunque la patogenia de la fibrosis sigue sin ser bien comprendida, hoy por hoy es evidente que la citoquina multifuncional TGF-β es esencial en el proceso. Existe una vía de señalización intracelular activada por el TGF-β; la transducción de señales comprende un sistema de receptores en la superficie celular y una familia de segundos mensajeros/factores de transcripción llamados Smads. Son proteínas evolutivamente conservadas. Los Smads son indispensables para la mayoría de las acciones profibróticas del TGF-β. Alteraciones en la actividad de los Smads conllevarían respuestas alteradas de la vía TGF-β con consecuencias catastróficas para el huésped. Los niveles de Smad 3 (activadores) están elevados en fibroblastos de la ES, no así los de Smad 4 o 7 (inhibidores).
Se desconoce el mecanismo por el que la vía del Smad de los fibroblastos está activada en la esclerodermia. La citada estimulación autocrina por parte del TGF-β endógeno podría ser una explicación.
El PDGF está ligado a la cicatrización de las heridas y a la fibrosis. Hay presencia de anticuerpos estimuladores contra el receptor del PDGF en el suero de pacientes con ES. El PDGF se deposita en vasos sanguíneos de la dermis profunda en lesiones precoces de ES y estimula la transformación de pericitos en fibroblastos.
ET-1. Actúa conjuntamente con el TGF-β en la conversión de fibroblastos en miofibroblastos. Los efectos beneficiosos de los inhibidores de los receptores de la ET-1 sobre la HAP nos llevan a deducir su importancia en la patogenia de la ES. La interleucina-4 tiene efecto profibrótico en fibroblastos de la dermis de ES. Se han implicado numerosas citoquinas en la angiogénesis, la angiostasis, la fibrosis y la inflamación localizada de la esclerodermia, aunque aún sin resultados concluyentes sobre las vías en las que participan.
Las moléculas de colágeno de las lesiones de ES muestran enlaces cruzados habituales en las moléculas del hueso, pero no presentes normalmente en la piel. Las fibrilinas 1 son fibrillas producidas por fibroblastos de la ES, más inestables que las de personas sanas. La interacción entre TGF-β y fibrilina es necesaria para la activación de los fibroblastos en la esclerodermia. Las integrinas α y β subregulan la síntesis de colágeno por parte de los fibroblastos, mecanismo que falla en la ES.
ROS. Se han implicado niveles altos de ROS y de estrés oxidativo en la esclerodermia. En la mayoría de las enfermedades inflamatorias, los altos niveles de ROS son consecuencia directa de la activación de células sanguíneas mononucleares. En la ES parecen ser independientes del estado inflamatorio e inducen daño en el ADN. Además, los radicales libres tienen efectos profibrogénicos en los fibroblastos. El PDGF estimula la producción de ROS.
Hipoxia. La hipoxia, frecuente en la piel de pacientes con esclerodermia, produce una activación de los fibroblastos, y una síntesis excesiva de ciertas citocinas estimuladoras, como el PDGF, el TGF-B1 y la endotelina. La transcripción de colágeno se ve aumentada cuando los fibroblastos se exponen a tensiones de oxígeno bajas, y esto puede deberse al TGF-β.
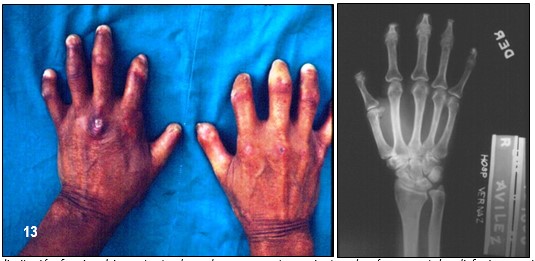
Imagen 13 con fines didácticos: Imagen izquierda: Severa esclerodactilia que ha deformado los dedos por retracción grave de la piel. Es evidente la pérdida espontánea de varias falanges distales. Ulcera crónica en el dorso de mano izquierda sugestiva de vasculitis. Estos cambios tróficos ocasionaban limitación funcional importante de ambas manos. La paciente además presentaba disfagia y episodios vasculares de Raynaud. Imagen derecha: Rx anterior de la mano derecha donde se evidencia reabsorción ósea severa de las falanges distales. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
– Otras manifestaciones clínicas: El Síndrome de Sjögren se presenta en el 20% de los pacientes y puede deberse a infiltración linfocitaria o sustitución fibrótica. Puede existir hipotiroidismo asociado a inflamación linfocitaria o sustitución fibrosa del tiroides. Se ha descrito neuropatía sensitiva del trigémino y de otros pares craneales, normalmente asociadas a Esclerosis Sistémica Limitada.
- Diagnóstico: Ante la sospecha clínica de ESP será necesaria la realización de una serie de exploraciones complementarias generales y otras encaminadas a descartar o establecer el grado de afectación orgánica. Desde la clínica, además de la exploración física general es necesario lleva a cabo una valoración del grado de afectación cutánea mediante escalas validadas, realizar una medición de la afectación articular e investigar el grado de afectación vascular a través de las características del fenómeno de Raynaud y de la presencia y características de úlceras isquémicas digitales.
Además de una analítica general, se realizará un estudio inmunológico que incluya determinación de anticuerpos antinucleares (ANA), anticuerpos anti-ENA (Ro, La, Sm, RNP), anticuerpos antitopoisomerasa I (Scl-70) y anticentrómero; VSG.
- Interpretación: Los ANA (patrón moteado o nucleolar), son positivos en el 90% de los casos. Dentro de las variantes del patrón moteado, se encuentran: El patrón moteado fino o discreto con cromosoma positivo. En este grupo se puede encontrar el patrón centrómero, característico de CREST que es una variante de ESP. El patrón moteado se encuentra también en algunos síndromes reumáticos no diferenciados. Cuando se encuentra un patrón moteado fino o moteado grueso, Si es cromosoma negativo, puede descartarse la presencia de anticuerpos Anti DNA. Los Anti Ro se pueden detectar en 9% y son indicativos de una enfermedad severa y rápidamente progresiva, incluyendo falla renal e hipertensión pulmonar. Los Anti U1-RNP se encuentran en un 6% y se asocian con la presencia de lupus eritematoso sistémico (LES), síndrome sobreposición o enfermedad mixta de tejido conectivo. Los anti Scl-70m se encuentra positivo en el 20-25%, y los anticentrómero en el 50%. La determinación del resto de anticuerpos descritos en la ESP (factor reumatoideo/FR, positivo en el 25% de los casos) no comporta significado diagnóstico ni pronóstico. De realización obligada son además, una videocapilaroscopia del lecho ungueal, una radiografía postero-anterior y de perfil de tórax y un ECG. Estudios posteriores para determinar el grado de compromiso orgánico son: estudio de la función respiratoria (espirometría), TAC pulmonar de alta resolución, y el aclaramiento endógeno de creatinina en orina de 24 horas.
La práctica de un tránsito esófago-gastro-duodenal así como una endoscopia digestiva (si existe clínica de reflujo gastroesofágico (RGE) significativo) es obligada para establecer la existencia y extensión de la posible afectación esofágica. La realización de una manometría esofágica queda reservada para los casos de alta sospecha de RGE con normalidad de las dos pruebas anteriores.
Para la valoración de la posible afectación pulmonar es imprescindible la práctica de pruebas de funcionalismo respiratorio, así como un scanner torácico de alta resolución.
La práctica de un ecocardiograma se reserva para aquellos pacientes con Esclerosis Sistémica Limitada de más de 10 años de evolución con el objetivo de descartar hipertensión pulmonar no asociada a afectación parenquimatosa, y en aquellos pacientes afectos de Esclerosis Sistémica Difusa a fin de valorar la posible afectación cardíaca. En este último supuesto, la realización de un SPECT de perfusión miocárdica puede ayudar a la valoración del grado de afectación del corazón. La práctica de radiografías simples de articulaciones supuestamente afectas nos ayudará a diferenciar el tipo de afectación articular. Para confirmar la existencia de calcinosis, será necesaria la práctica de radiologías de partes blandas.

Imagen 14 con fines Didácticos: El patrón moteado fino se caracteriza por tinción del núcleo con gránulos finos o gruesos, los nucléolos no se tiñen así como tampoco se tiñe la placa de la cromatina en células en división. Imagen 15 con fines didácticos: Test de Schirmer: esta prueba determina si el ojo produce suficientes lágrimas para mantenerse húmedo. Resultado normal: longitud menos de 10 mm de humedad en papel filtro en 5 minutos. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.
El fondo de ojo y la ecografía doppler renal son pruebas de realización inicial ante la sospecha de una crisis renal esclerodérmica. La práctica de pruebas invasivas como la angiografía renal y la punción-biopsia renal están indicadas cuando haya duda diagnóstica ante la gravedad del cuadro.
La existencia en la analítica de una elevación de la creatincinasa (CPK/MM) y aldolasa muscular pueden indicar la existencia de una miopatía asociada. En dicho caso y en función del grado de debilidad, estaría indicada la realización de un electromiograma y de una biopsia muscular.
Finalmente, si existe clínica de síndrome seco se realizarán las pruebas de Schirmer y Rosa de Bengala para descartar un síndrome de Sjögren asociado, que de ser positivas, deberían apoyar la realización de otras pruebas conducentes a establecer el codiagnóstico.
Otros exámenes pueden abarcar:
- – Radiografía de tórax. Tomografía computarizada de los pulmones de alta resolución. Ecocardiografía.
- – Análisis de orina/clearance endógeno de creatinina.
- – Pruebas de funcionamiento pulmonar (espirometría) y digestivo.
- – Biopsia de piel.
Curso y pronóstico.- La historia natural de la ESP es variable. La sobrevida a 10 años del diagnóstico es de 65%.

Terapia.- (Debe ser dirigida al órgano comprometido). No existe tratamiento específico. Se usan: Corticosteroides, Inmunodepresores, (AINES), etc. No existe prevención.
comprometido). No existe tratamiento específico. Se usan: Corticosteroides, Inmunodepresores, (AINES), etc. No existe prevención.
La evaluación de las terapias en la ESP no ha sido una tarea fácil. Se ha usado d-penicilamina, un inmunomodulador que interfiere en el cross-linking del colágeno. Disminuye la fibrosis de la piel y retarda la progresión del compromiso cutáneo en algunos enfermos. Puede estabilizar la progresión de la fibrosis pulmonar.
- -Fenómeno de Raynaud: Abstenerse de fumar y de exponerse a ambientes fríos. Uso de nifedipino como vasodilatador periférico que relaja la musculatura lisa vascular; el prazosin también ha demostrado su utilidad. En casos de Raynaud grave con ulceras isquémicas, pueden utilizarse prostaciclina o análogos en infusión intravenosa intermitente. La prostaciclina oral no se ha demostrado ser eficaz. Recientemente se ha demostrado la utilidad de fármacos inhibidores de los receptores de la endotelina (Bosentán), pudiendo prevenir la aparición de nuevas ulceras, aunque no ha demostrado eficacia en la curación de las mismas. Los inhibidores de la fosfodesterasa-5, como el sildenafilo, también parecen mejorar tanto el Raynaud como las ulceras isquémicas, aunque falta estudios controlados. Otros vasodilatadores como la reserpina, también pueden usarse. El bloqueo simpático y la simpatectomía pueden ser útiles, pero suele perder eficacia con el paso del tiempo.
- – La hipertensión arterial: así como la afectación renal, se intentará controlar con antihipertensivos tipo IECA.
- – Úlceras de los dedos: con vendaje compresivo local. Evitar la humedad y secarlas al aire. Si se infectaran hacer uso de antibióticos orales o locales y desbridamiento. En lo posible no amputar.
- – Artritis y/o artralgias: con AINE o dosis bajas de prednisona (<10 mg/día). Ejercicios suaves de estiramiento para disminuir contracciones. Fisioterapia y rehabilitación.
- – Dismotilidad esofágica/gástrica o colónica: La disfunción esofágica mejora con medidas posturales luego de las comidas principales como medida antireflujo, así como otras como el uso de, bloqueantes H2 (ranitidina, p. ej.), inhibidores de la bomba de protones (omeprazol, p. ej,), y pro cinéticos (Mosapride, domperidona, Metoclopramida, p. ej). El vaciamiento gástrico puede mejorar con pro cinéticos (Metoclopramida), y eritromicina. El octreotido subcutáneo estimula la motilidad intestinal y disminuye el sobrecrecimiento bacteriano. Para la malabsorción puede utilizarse antibióticos de amplio espectro.
- – Enfermedad Pulmonar: en la fibrosis intersticial en etapa inicial con infiltración celular activa se ha usado esteroides y ciclofosfamida oral, o con ciclofosfamida IV seguida de azatioprina oral, con resultados variables. Ningún tratamiento es eficaz en la fibrosis pulmonar establecida.
- – Hipertensión pulmonar: Al igual que para la hipertensión pulmonar (HTP) primaria, en los casos con respuesta positiva al test vasodilatador agudo, realizado durante el cateterismo derecho necesario para confirmar el diagnóstico de HTP, puede utilizarse bloqueantes del calcio, con buena respuesta a largo plazo y disminución de la mortalidad. La respuesta al test agudo es habitualmente negativa en los pacientes con HTP asociada a ESP. El uso de iloprost (prostaciclina) como vasodilatador ha sido reportado de algún beneficio. En algunos enfermos se ha realizado con éxito transplante corazón- pulmón.
Diferentes fármacos utilizados en la ESP de acuerdo al órgano afectado:
- – Vasodilatadores: Fenómeno de Raynaud: Nifedipino, verapamilo. Losartan. Iloprost
- – Hipertensión pulmonar: Epoprostenol. Iloprost (carboprostaciclina). Captopril
- – Crisis renal: Captopril, Enalapril. Diálisis renal. Trasplante renal. Inmunosupresores
– Endurecimiento de la piel: D-penicilamina. Colchicina. Interferón gamma. Relaxina. Metotrexate. Ciclosporina. Agentes antifibroticos. La más importante es la D-penicilamina, la cual reduce la participación cutánea y mejora la sobrevida a 5 años si se empieza el tratamiento en etapas tempranas de la enfermedad. Sin embargo, también presenta varios efectos secundarios. – Enfermedad pulmonar intersticial: Ciclofosfamida. Antifibróticos.
Complicaciones.- La causa más común de muerte en personas con esclerodermia es la cicatrización de los pulmones, denominada fibrosis pulmonar.
Otras complicaciones de la ESP abarcan:
- – Cáncer.
- – Insuficiencia cardíaca.
- – Presión arterial alta en los pulmones (hipertensión pulmonar).
- – Insuficiencia renal.
- – Problemas para absorber los nutrientes de los alimentos (malabsorción).

Imagen 108.- ESP variante C.R.E.S.T. La piel alrededor de la boca está tirante y la paciente la abre tanto como ha podido. Telangiectasia en el dorso de la nariz. La piel posee un aspecto brillante y atrófico. En la variedad limitada con calcinosis, fenómeno de Raynaud, esclerodermia, alteración esofágica y telangiectasias, denominada CREST por sus siglas, suelen ser positivos los anticuerpos anticentrómero. Imagen 109-110.- ESP. variante C.R.E.S.T. Severa esclerodactilia que ha deformado los dedos, varios de ellos en riesgo de evolucionar a la gangrena. La paciente presentaba episodios vasculares de Raynaud. Son evidentes las vasculitis de las manos y dedos, con lesiones en “mordida de rata”, en la punta de los dedos. Imagen 111.- ESP. variante C.R.E.S.T. Calcinosis discreta en la punta del dedo índice, la misma que se hace evidente en la radiografía simple del dorso de las manos. Los cambios acrales se debieron a crisis de Raynaud. Imagen 112 a-b-c.- ESP variante C.R.E.S.T. En A no se observa lesión ósea. Hay imágenes calcificadas en los tejidos blandos en la región del codo. En B, dilatación del esófago y ausencia casi total de peristaltismo, a nivel de una moderada hernia de hiato. Obsérvese alteraciones intersticiales del parénquima pulmonar, graves y bilaterales, en los lóbulos inferiores. En C, extensos depósitos de colágeno denso en la dermis con aparente ausencia de apéndices y adelgazamiento de la epidermis. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo I, 2018, UEES, Ecuador.
Pronóstico.- La historia natural de la enfermedad es variable. La mayoría de los enfermos, sin embargo, tiene un curso prolongado Las formas difusas tienen peor pronóstico que las limitadas, porque presentan con mayor frecuencia afectación renal, pulmonar y/o cardíaca. Por otro lado, hay pacientes con esclerodermiaque pueden desarrollar Cirrosis Biliar Primaria (CBP), estos suelen presentar la forma cutánea limitada de esclerodermia. Por otra parte, la asociación de otras enfermedades autoinmunes en pacientes con CBP se ha descrito hasta en un 60% de los casos, de las cuales, el síndrome CREST constituye hasta un 15% del total. La asociación de síndrome CREST y CBP ha merecido una denominación propia (síndrome de Reynolds) y según algunos autores representa un subgrupo diferente de pacientes con características clínicas bien diferenciadas y un mejor pronóstico.
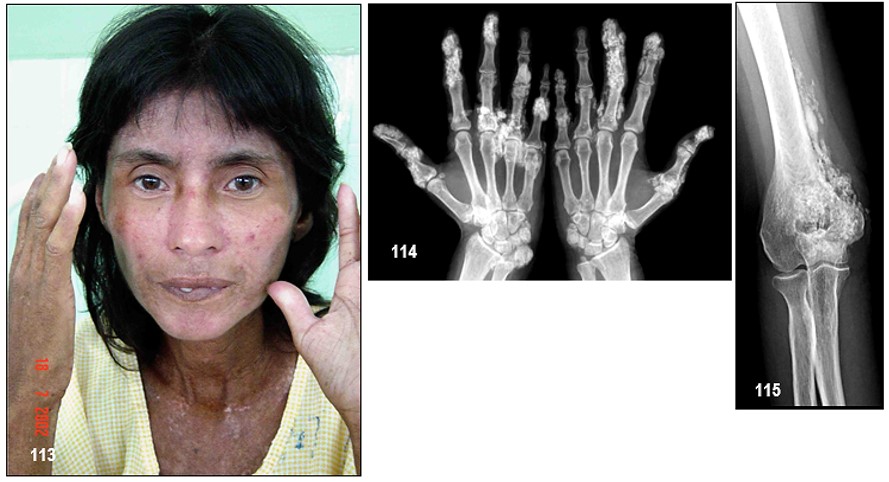
Imágenes 113-114-115.- C.R.E.S.T.– Clásicas imágenes paradigmáticas de “facies de máscara” del C.R.E.S.T. Cara, piel tensa y atrófica, con labios apretados y telangiectasias enmarañadas y diseminadas en toda la cara. Es evidente la desaparición de las líneas de expresión de la cara, lo que le brinda una apariencia inexpresiva pero más joven a la edad real. Signos de vasculitis en las manos (algunas úlceras pequeñas en artejos) e hipopigmentación cutánea (en sal y pimienta) en cuello y parte superior del tórax. La paciente presentaba depósitos de calcio (calcinosis cutis) en las puntas de los dedos de ambas manos, así como acumulus de calcio en las articulaciones de los codos (Calcinosis cutis), fenómeno de Raynaud, disfagia Esofágica (de Esophagus, de su nombre en inglés), Sclerodactilia (de su nombre en inglés) y Telangiectasias. Para colmo la paciente desarrolló fibrosis pulmonar intersticial. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo I, 2018, UEES, Ecuador.
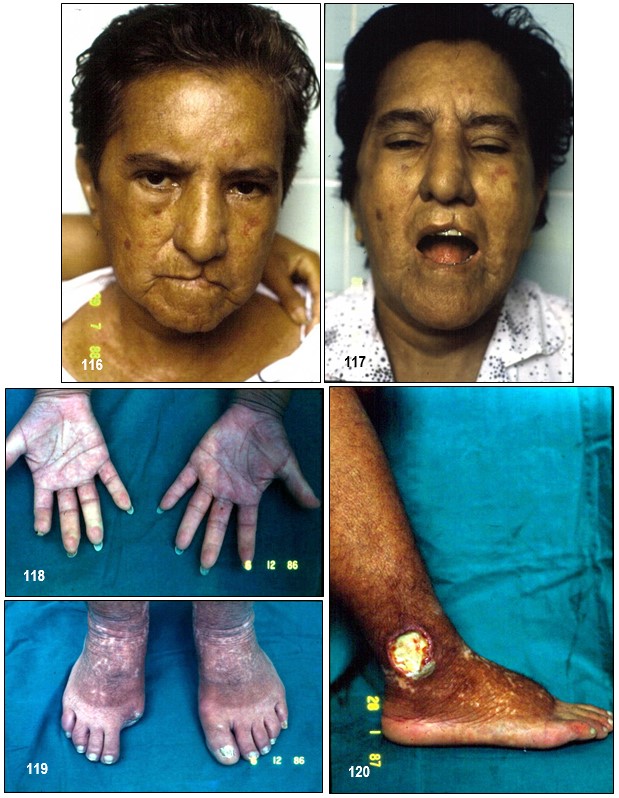
Imagen 116.- ESP variante C.R.E.S.T. Rostro con evidente compromiso esclerodérmico; véanse los pliegues perilabiales en “bolsa de marinero”, además de telagiectasias. Imagen 117.- ESP variante C.R.E.S.T. Imposibilidad para abrir completamente la boca. Se solicitaron anticuerpos antinucleares y anticentrómero; radiografías. Imagen 118.- C.R.E.S.T. Severa esclerodactilia que ha deformado los dedos con pérdida espontánea de la falange distal del dedo índice de la mano derecha. Varios de ellos en riesgo de evolucionar a la gangrena. La paciente presentaba episodios vasculares de Raynaud. Aquejaba igualmente disfagia para sólidos. Imagen 119.- C.R.E.S.T. La gangrena debido a vasculopatía obliterante ha ocasionado amputación del dedo gordo. Imagen 120.- C.R.E.S.T. úlcera en la zona del “calcetín”, de aspecto vasculíticas. La localización en el talón es excepcional. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo 1. 2018, UEES.

Imagen 121.- ESP variante C.R.E.S.T. Esclerodactilia con pérdida espontánea de la falange distal por reabsorción ósea, fenómeno de Raynaud en etapa blanca. Imagen 122, ESP variante C.R.E.S.T.- nótese que la radiografía correspondiente a la mano afectada evidencia acrosteolisis de la falange distal. Calcificación discreta en el primero, segundo y tercer dedo. Rarefacción ósea. Imágenes 123-124.- ESP variante C.R.E.S.T. Inmunofluorescencia indirecta. En 8, anticuerpos antinucleares positivo (ANA) (x 500). Anticuerpos anticentrómero positivo (AAC) (x 800). Los ANA se encuentran, aproximadamente, en el 90% de los pacientes con esta entidad, siendo el patrón nucleolar el más característico de la esclerodermia; otro anticuerpos el denominado anticuerpo contra el antígeno nuclear soluble Scl-70 es específico de la esclerodermia, pero se encuentra solamente en cerca de un 25% de pacientes con esta entidad. Con relación al AAC está asociado con esclerodactilia en la variante CREST. Fuente: Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo I, 2018, UEES, Ecuador.
Comentarios de los autores: La presencia de telangiectasias en fieltro (véase la imágenes 113 y 116), a veces son signos de comienzo de la enfermedad, de gran valor diagnóstico, algunas de contornos cuadrangulares en una paciente con síndrome de CREST o síndrome de Thibierge-Weissenbach, son muy promisorias para un certero diagnóstico. La imágenes 111-114-115 muestra la calcinosos cutis a nivel de los dedos de ambas manos, aunque pueden presentarse en otros sitios, en especial en áreas de extensión (antebrazos (imagen 115).
INFORMACIÓN BÁSICA: Síndrome C.R.E.S.T (ORPHA: 220393). (Calcinosis, fenómeno de Raynaud, alteraciones de la motilidad Esofágica, Sclerodactilia y Telangiectasias) es una forma limitada de la Esclerosis Sistémica Progresiva en la que existe depósito de cristales de hidroxiapatita (calcio) como resultado de un trastorno localizado del tejido conectivo de la piel, bolsas serosas, tendones, cápsula articular, membrana sinovial y cartílago (calcificación “distrófica: Imagen 4A”). Los cristales de hidroxiapatita forman parte del hueso normal y también son los responsables de la mayoría de las calcificaciones de partes blandas del organismo. En esta modalidad de la entidad es típico que el síndrome de Raynaud anteceda en años a la presentación del resto de los síntomas de enfermedad. Además de las manifestaciones clásicas de la piel pueden aparecer manifestaciones de fibrosis intersticial pulmonar que se evidencia clínicamente por estertores húmedos bibasales muchas veces sin otra forma de expresión, esto causa hipertensión pulmonar y puede desencadenar una cardiopatía pulmonar hipertensiva con las consecuentes manifestaciones de insuficiencia cardiaca derecha. Se señala como frecuente la asociación del síndrome de C.R.E.S.T con la cirrosis biliar primaria.
Patogenia.- Es de patogenia auto inmune heterogénea, se señala que existen clones de células T dominantes cuya disfunción puede dar origen a las colagenosis en general y a esta en particular, además de la característica disfunción de los linfocitos B. Existen anticuerpos antinucleares y anticuerpos anti centrómeros que son aún más frecuentes en el síndrome de C.R.E.S.T. Los componentes del C.R:E.S.T. son:
Manifestaciones clínicas.-
- – Calcinosis (calinosis cutis). Se caracteriza por la formación de depósitos de calcio (nódulos) en la piel de manos y rostro, mismos que pueden ser dolorosos y romperse, volviéndose visibles como material blanco y susceptible a sufrir infección.
- –Fenómeno de Raynaud (Raynaud’s phenomenon). Es el síntoma más común y característico (se presenta en 70 % de los pacientes) y es una alteración de la circulación como respuesta al estrés o al frío, que genera cambios en el color de la piel: blanco debido a reducción en la afluencia sanguínea, azul cuando la parte afectada pierde oxígeno, y rojo en el momento en que la situación se empieza a normalizar. Este proceso es más patente en pies y manos, pero también suele aparecer en lengua y rostro; llega a ser doloroso y genera paulatino engrosamiento cutáneo en zonas afectadas, así como falta de expresión.
- – Disfunción del esófago (esophageal hypomotility). Los pacientes pueden desarrollar irregularidades en todo el sistema digestivo debido a que la sobreproducción de colágeno crea fibrosis. Resulta especialmente afectada la parte baja del esófago, donde las lesiones impiden que el cardias se cierre adecuadamente para evitar el reflujo; como resultado, los ácidos gástricos son expulsados, causando esofagitis por reflujo, esto lleva a lesiones cicatriziales que estrechan el esófago, generando la disfagia.
- – Esclerodactilia (sclerodactyly). Es el endurecimiento de los artejos de los dedos tanto en pies como en manos; generalmente aparece luego de que se padece inflamación de la piel. Se caracteriza porque los dedos adquieren apariencia tensa y brillante, además de que pueden verse afectada su flexibilidad, volviéndose curvos en una posición similar a una garra (contracturas).
- – Telangiectasia (telangiectáses). La formación de depósitos de calcio en piel y tejidos subcutáneos hace que las venas localizadas en mejillas, contorno de los labios y dedos se dilaten de manera anormal, por lo que adquieren la apariencia de ramificaciones de color morado, también conocidas como telarañas vasculares o telangiectasias.
Existen diversas teorías acerca de por qué se presenta la sobreproducción de colágeno que genera el síndrome de C.R.E.S.T. Una de ellas sugiere que el propio sistema inmunológico (que defiende al organismo de infecciones) juega papel fundamental. Esto se deduce a través de estudios de sangre en los que se revela alto índice de citokinas, sustancias que crea el organismo para coordinar a las defensas del cuerpo contra bacterias, virus y otros invasores extraños. Como resultado, las defensas del organismo atacan a los tejidos sanos del propio cuerpo y estimulan la sobreproducción de colágeno. Otra hipótesis sugiere que el padecimiento se debe a que los vasos sanguíneos, especialmente los pequeños (capilares), se estrechan y endurecen de manera sobre exagerada ante estímulos de frío o estrés; dicha respuesta, originada por infecciones o características genéticas, causa deficiente distribución de sangre en órganos y tejidos de regiones determinadas de la piel, les genera lesiones y, por tanto, comienza a producirse exceso de colágeno.
Actualización (24 Octubre 2023): El mayor estudio genómico de Raynaud hasta la fecha
Los investigadores revisaron los registros médicos electrónicos del Biobanco del Reino Unido, una base de datos a gran escala que contiene información genética y de salud de medio millón de participantes. Identificaron a más de 5,100 individuos con la enfermedad de Raynaud, de los cuales el 68 por ciento tenían la enfermedad de Raynaud primaria. Estos participantes se compararon con más de 439,000 controles que no tenían la enfermedad de Raynaud. En un análisis secundario, el equipo también utilizó registros de salud del Estudio de Genes y Salud de la Universidad Queen Mary de Londres, que contiene información de salud sobre individuos de ascendencia del sur de Asia.
Los investigadores identificaron dos genes que probablemente estén involucrados con la enfermedad de Raynaud. El primero, ADRA2A, codifica para el receptor adrenérgico alfa-2A que puede causar vasoconstricción de pequeños vasos sanguíneos en respuesta a las hormonas del estrés. Los investigadores han sospechado durante mucho tiempo que este tipo de receptor podría estar involucrado con la enfermedad de Raynaud, pero hubo un debate sobre qué subtipo de receptor era el responsable.
«Nuestro hallazgo de los receptores alfa-2A es bastante interesante porque el enfoque siempre ha estado en los receptores alfa-2C», dijo el Dr. Pietzner. «Es solo una carta, pero es una diferencia enorme en términos de biología y fisiología», dijo, y podría ser la razón por la que las terapias dirigidas a los receptores 2C han sido ineficaces.
La segunda asociación más fuerte fue para el factor de transcripción IRX1. Se sabe menos acerca de este gen, pero los datos que tenemos sugieren que está involucrado en la regulación de la dilatación de los vasos sanguíneos, señaló el doctor Pietzner.
«Podría haber un equilibrio entre el hallazgo de ADRA2A como responsable de la constricción y el hallazgo de IRX1 indirectamente relacionado con la dilatación de esos vasos después de las constricciones. Tener ambos puede explicar por qué estos episodios prolongados de vasoconstricción conducen a una pérdida de oxígeno en los tejidos», por lo que se vuelven blancos y luego azules, dijo.
Debido a que la cohorte del Biobanco estaba centrada en Europa, el Dr. Pietzner y sus colegas también identificaron 400 casos de Raynaud en individuos británicos de ascendencia bangladesí y paquistaní y pudieron replicar la asociación entre IRX1 y Raynaud. No se disponía de datos sobre ADRA2A.
Los genes identificados están asociados con la enfermedad de Raynaud primaria. La enfermedad de Raynaud secundaria es un tipo más raro de la afección que se presenta junto con trastornos autoinmunes, como la esclerodermia, y generalmente es más grave.
Durante mucho tiempo se ha sospechado que la enfermedad de Raynaud tenía algún componente genético, porque la mitad de los pacientes con la enfermedad de Raynaud tienen otro miembro de la familia con la misma afección, dijo la doctora Laura Hummers, codirectora del Centro de Esclerodermia John Hopkins en Baltimore. Ella no participó en el estudio.
Este es «el estudio más grande de este tipo que se ha hecho», dijo, y el primero en mostrar un mecanismo potencial detrás de esta asociación genética.
El principal hallazgo genético, ADRA2A, «apunta a un receptor en las células que regula el tono de estos vasos sanguíneos», continuó. «Sugiere que tal vez hay demasiados de estos receptores o que son demasiado sensibles; Hay algo diferente en ellos que hace que los pacientes sean más susceptibles a estos desencadenantes del resfriado. Saber eso es potencialmente muy importante, porque podría darte una forma directa de intervenir, si es cierto».
Diagnóstico.- Es el propio aplicado para la Esclerosis Sistémica Progresiva (véase ESP, en este mismo apartado).
Bibliografía
- Vasudevan AR, Ginzler EM. Clínicasl features of systemic lupus erythematosus. En: Rheumatology. Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (ed). Mosby Elsevier (2011).
- Hahn BH. Lupus eritematosos sistémico. En: Harrison. Reumatología. Fauci AS, Langford CA (eds). Ed. MCGraw Hill-Interamericana (2007).
- Tama Viteri FA; Barberán Torres PJ. Atlas y Texto en Color de Imágenes Clínicas, Tomo I, 2018, UEES, Ecuador.
- Ferrandiz, C.: «Dermatosis eritematoescamosas (I). Psoriasis. Eritrodermias.» en Dermatología clínica.- Madrid: MMI Elsevier España, S.A. ISBN 84-8174-537-5
- American College of Rheumatology 2012
- Fitzgerald O. In: Psoriatic arthritis. Firestein GS, Budd RC, Gabriel SE, et al, eds. Kelley’s Textbook of Rheumatology. 9th ed. Philadelphia, Pa: Saunders Elsevier; 2012: chap 77.
- Husni ME. Psoriatic arthritis. In: Carey WD, ed. Cleveland Clinic: Current Clínicasl Medicine. 2nd ed. Philadelphia, Pa: Saunders Elsevier; 2010.
- http://www.reumatologiaclínicas.org/
- http://www.ser.es/wp-content/uploads/2015/10/Manual-SER.pdf
- Investigadores relacionan dos genes con la enfermedad de Raynaud | MDedge Reumatología