.
INFORMACIÓN BÁSICA.- Encondromatosis Múltiple Familiar (OMIM:166000/ORPHAN:296 /CIE-10:Q78.4). La Osteocondromatosis Familiar Múltiple o Encondromatosis Múltiple Familiar (EMF) se caracteriza por el desarrollo de 2 o más osteocondromas, que son tumores cartilaginosos benignos, que se localizan principalmente cerca de las metáfisis de los huesos largos, y están formados por hueso medular y cortical recubierto de una capa de cartílago hialino. Crece exterior a la metáfisis, actuando como un platillo de crecimiento ectópico, que cesa su crecimiento a la maduración del esqueleto.
La EMF no es una enfermedad frecuente. Aunque suele manifestarse en la primera década de vida la consulta suele ser tardía, bien porque en la familia alguien la presenta y conoce la naturaleza benigna de la enfermedad, que, en muchas ocasiones, no produce molestias al paciente o bien porque, al ser lesiones que se localizan fundamentalmente en las metáfisis, suelen hacerse evidentes y manifestarse clínicamente con cada brote de crecimiento y tienden a estabilizarse al final del desarrollo.
Los encondromas se localizan principalmente en huesos largos aunque no es infrecuente hallarlos en huesos planos. Las manifestaciones clínicas son variadas, desde casos asintomáticos o simples deformidades estéticas, hasta limitación de la función articular o tendinosa con alteración del crecimiento en los niños o, incluso, grandes deformidades que producen síntomas por compresión de estructuras vecinas. Aunque generalmente son lesiones benignas, la complicación más importante es la degeneración maligna a osteocondrosarcoma que siempre se sospechará en caso de crecimiento rápido de una lesión previa y/o aparición de dolor. En estos casos, la cirugía es la piedra angular del tratamiento junto a la quimioterapia pre y post-operatoria.
La EFM tiene diferentes nominaciones: aplasia diafisaria, osteocondromatosis múltiple hereditaria, exostosis múltiple hereditaria, enfermedad exostosante múltiple, exostosis cartilaginosa múltiple hereditaria o condrodisplasia deformante hereditaria, aclasia diafisaria. Entidad descrita en 1891 por Bessel-Hagen, es una enfermedad poco frecuente que se hereda con un rasgo genético autosómico dominante con penetrancia variable, aunque se han descrito casos de herencia recesiva e inclusive esporádicos. Se han identificado tres loci en relación con la enfermedad: EXT1 brazo largo del cromosoma 8q23-8q24 (responsable del 60-70% de los casos); EXT2 brazo corto del cromosoma 11p11-11p12 (20-30% de los casos) y EXT3 brazo corto del cromosoma 19p. La EFM se ha descrito asociada al síndrome de Langer-Gienon, la leucemia mieloide aguda, la espondilitis anquilopoyética o el síndrome de Down.
Epidemiologia.- La prevalencia de la EFM oscila entre 1/20.000-50.000 habitantes, estimándose que en España hay alrededor de 500 familias afectadas, desconociéndome en nuestro medio este dato, quizás esta descripción sea la primera denunciada. La EFM tiene una clara predilección por el sexo masculino (2:1) y es en este en el que se presentan las manifestaciones clínicas más graves. Los osteocondromas de la EFM se pueden localizar en casi todo el esqueleto adoptando una distribución simétrica y aunque predominan en las metáfisis tanto proximales como distales de huesos largos, no es infrecuente hallarlos en huesos planos.
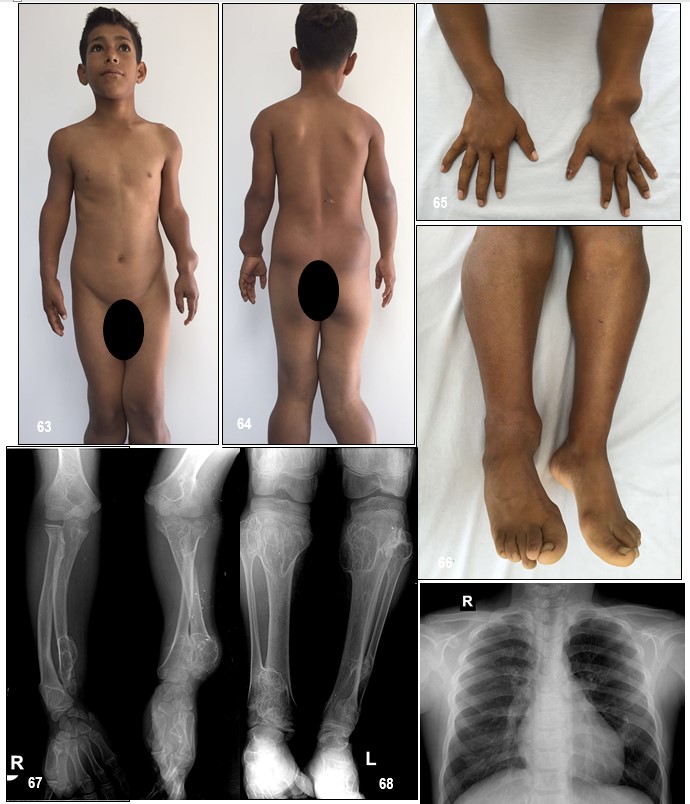
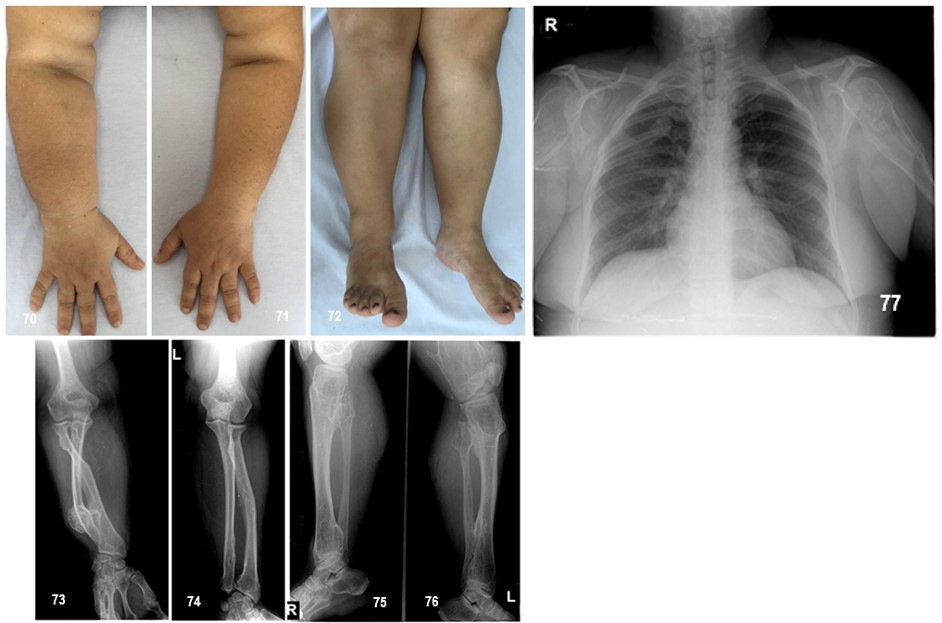
Manifestaciones clínicas.- Los primeros osteocondromas se presentan durante la primera década de la vida y, a diferencia de otras encondromatosis, una vez que se cierran los cartílagos del crecimiento se detiene la formación de nuevos encondromas. Las malformaciones musculoesqueléticas más frecuentes son: el acortamiento del cúbito con arqueamiento del radio (39-60%), crecimiento asimétrico de las extremidades (10-50%), baja estatura (37-44%), angulación en varo o valgo de la rodilla (8-33%) y deformidad de tobillo (2-54%).
En fin, las manifestaciones clínicas son variadas; desde casos asintomáticos o simples deformidades estéticas, a deformidades que producen síntomas por compresión de estructuras vecinas tanto en partes blandas, nervios periféricos o vasos, hasta importantes limitaciones de la función articular o tendinosas que pueden producir alteraciones en el crecimiento de los niños.
Las malformaciones musculoesqueléticas más frecuentes son: acortamiento del cúbito con arqueamiento del radio, asimetría de extremidades, baja estatura, angulación en varo o valgo de la rodilla y deformidades del tobillo. El diagnóstico de la EFM se establece por la sospecha clínica y se apoya en los estudios de imagen y los antecedentes familiares.
Los osteocondromas se desarrollan y aumentan de tamaño durante la primera década de la vida y dejan de crecer cuando las placas de crecimiento se cierran durante la pubertad.
Los tipos de condromas pueden ser pediculados o sésiles (base ancha) aunque predominan éstos últimos, y su tamaño puede variar mucho.
La localización de los condromas en los huesos que se desarrollan a partir de cartílago, especialmente en los huesos largos de las extremidades. Las más comunes son el fémur proximal o distal, húmero proximal, tibia proximal, radio distal, pelvis, y escápula. Son particularmente frecuentes alrededor de la rodilla, donde tiene más probabilidades de sufrir una degeneración maligna.
Los huesos faciales no están afectados; las manos, en cambio, sí suelen afectarse. La distribución suele ser simétrica y bilateral, aunque se han descrito casos con compromiso unilateral.
Se ha visto que el grado de afectación y deformidad de antebrazos y tobillos es una medida de la extensión global de la enfermedad. Un mayor número de exostosis, deformidades angulares prominentes, y acortamiento en estas áreas, indica afectación esquelética más severa y generalizada.
El porcentaje de osteocondromas sésiles se correlaciona con la extensión de las deformidades, con deformidades angulares más graves cuando más del 90% de los osteocondromas son sésiles. La Tabla 6 resume las manifestaciones clínicas.

Complicaciones.- La complicación más grave es la degeneración maligna hacia osteocondrosarcoma. Véase la Tabla 7.

Se estima que la transformación sarcomatosa de los encondromas solitarios ocurre en menos del 1%, pero en los pacientes con EFM el riesgo se incrementa hasta el 25% hacia los 40 años de edad. Las localizaciones más frecuentes de degeneración maligna son la pelvis, la cadera y la cintura escapular. Siempre se sospechará en los casos de aparición de dolor y/o crecimiento rápido de las tumoraciones y cuando los hallazgos radiológicos sean característicos: espesor del cartílago superior a 2cm en adultos o 3cm en niños o rotura de la cortical y extensión a partes blandas adyacentes. En estos casos, la técnica diagnóstica de elección es la RM que ofrece una sensibilidad muy superior a la tomografía computarizada (TC). Es importante resaltar que ni la historia clínica ni la exploración física ni los estudios complementarios bastan por sí solos para determinar el carácter benigno o maligno de una lesión ósea por lo que, en los casos de alta sospecha de malignización, se realizará biopsia de aquélla. La biopsia por punción-aspiración mediante aguja fina (PAAF) puede proporcionar resultados falsos negativos, por lo que se recomienda la biopsia escisional, que posee la ventaja de proporcionar suficiente material que disminuya el riesgo de error. Debe completarse el estudio de extensión con TC toracoabdominal y tomografía por emisión de positrones (PET).
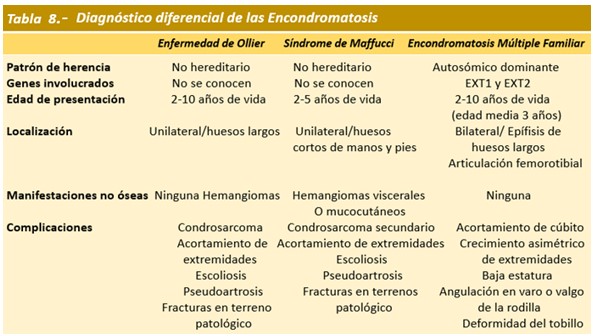
Diagnóstico.- En la infancia, alrededor de los 5 años, la mayoría antes de los 12 años, generalmente por masa palpable o deformidad con angulación de un hueso. La existencia de familiares afectados, la bilateralidad de las lesiones, su localización en los huesos largos y el cese de crecimiento de las lesiones al finalizar el desarrollo orientan al diagnóstico de EFM. La localización unilateral, ausencia de familiares afectados y localización más frecuente en manos apoyan el diagnóstico de la enfermedad de Ollier y la unilateralidad y presencia de hemangiomas viscerales y/o mucocutáneos a la enfermedad de Mafucci.
Podríamos concluir diciendo que el diagnostico se basa en la documentación clínica y radiográfica, complementada, si es posible, con un análisis histológico de los osteocondromas. La radiología convencional permite diagnosticar la enfermedad siendo las imágenes características.
Diagnóstico diferencial.- El diagnóstico diferencial se planteará con la displasia epifisaria heminélica, los osteomas, los condromas periósticos, por lo general únicos y, especialmente, con otras encondromatosis múltiples: la enfermedad de Ollier y el síndrome de Mafucci. La existencia de familiares afectados, la bilateralidad de las lesiones, su localización en los huesos largos y el cese de crecimiento de las lesiones al finalizar el desarrollo orientan al diagnóstico de EFM, como en los casos que describo. La localización unilateral, ausencia de familiares afectados y localización más frecuente en manos apoyan el diagnóstico de la enfermedad de Ollier y la unilateralidad y presencia de hemangiomas viscerales y/o mucocutáneos a la enfermedad de Mafucci; véase la tabla 8.
Tratamiento.- La conducta terapéutica está estrechamente relacionada con la sintomatología clínica, variando desde una actitud meramente observacional y conservadora hasta el tratamiento quirúrgico. Este puede abarcar desde la resección del tumor con osteotomías correctivas en los casos más leves, hasta la amputación del miembro afectado en las formas más agresivas e incapacitantes. No existe un tratamiento curativo para la OMH y solo estaría indicado el tratamiento de aquellas exostosis que provocasen deformidades estéticas, trastornos funcionales o dolor y, salvo casos excepcionales, se aconseja posponer la intervención hasta después de finalizar el período del crecimiento con el fin de evitar recidivas.
Conclusiones.- La EFM es una enfermedad hereditaria que se manifiesta en la primera y segunda décadas de la vida, con clara predilección por el sexo masculino. Los encondromas se localizan fundamentalmente en las metáfisis de los huesos largos, crecen mientras el esqueleto es inmaduro y dejan de crecer al finalizar el desarrollo. De carácter benigno, la posibilidad de malignización y el avance de sus deformaciones requieren controles clínicos y radiológicos periódicos con el fin de detectar precozmente sus complicaciones. Se debe aconsejar estudio genético a los familiares de los pacientes afectados.
En la encondromatosis El riesgo de malignización en la encondromatosis es superior a 10%, hasta 30% en algunas series, especialmente en las lesiones de ubicación distal, y hasta 40% cuando son múltiples y axiales.
Es importante considerar lo anterior cuando se toma la decisión de intervenir en el curso de esta enfermedad.
Bibliografía
- E.L. Sabadotto, A.O. ansur. Exostosis cartilaginosa múltiple. Arch Argent Pediatr, 98 (2000), pp. 388-392Carpintero, J.A. Del Fresno, R. Carpintero, M.J. Gálvez, M.A. Marín. Complicaciones de los osteocondromas Rev Esp Cir Osteoar, 237 (2009), pp. 22-29
- López Roldán, S. Alvarez Gómez, J.L. González Pulido. Actualización del osteosarcoma para el médico de familia. Semergen, 37 (2011), pp. 22.
- Asociación Española de Osteocondromas Múltiples Congénitos (AEOMC). Introducción a HME/OMC. (Acceso: 07-10-15). Disponible en: http://aeomc.blogspot.mx/2007/05/introduccin-hmeomc.html.
- J.A. Sansón-Rio Frío, S. Navarro Santiesteban, R.I. Bahena, V.V. Villavicencio, H. Martinez-Said, R.A. Padilla. Encondromatosis múltiple familiar, diagnóstico diferencial: presentación de un caso clínico con condrosarcoma y revisión de la literatura. Act Ortop Mex, 23 (2009), pp. 376-382
- Álvarez Cambras, O.M. de Cárdenas Centeno, L.O. Marrero Riverón, R.D. González Cabrera, A. Tamayo Figueroa, V. Rondón García. Condrosarcoma secundario de la pelvis: presentación de dos casos Rev Cubana Ortop Traumatol, 10 (1996), pp. 168-173
- Rambeloarisoa, M. Guedy, M.L. Legeai. Hereditary multiple exostoses alter 40 years of development: a case report. Rev Med Interne, 23 (2002), pp. 657-664
- Gómez-Valencia, A. Morales-Hernández,J. Salomón-Cruz, A.J. Berttolini Diaz, R.M. Cornelio-García,E. Toledo-Ocampo. Exostosis múltiple hereditaria y síndrome de Down. Bol Med Hosp Infant Mex, 62 (2005), pp. 356-361.
- García-Lamazares, E. Someso-Orosa, A. Zamora-Casal, E. Rodríguez-Moldes. Osteocondromatosis múltiple familiar: a propósito de un caso. Semergen 2013;39:171-4
- Orphanet: Síndrome de Langer Giedion
- www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=502
- J.V.M.G. Bovee. Multiple osteochondromas. Orphanet Journal of rare diseases, 3 (2008), pp. 3
- Muñoz Villa. Tumores óseos. Pediatr Integral, 12 (2008), pp. 693-700
- R.C. Pacheco López, B. Torres Gómez, J.A. Ugalde Vitelly, C. del Vecchyo Calcaneo, N. Sastré Ortiz. Enfermedad de Ollier de presentación bilateral. Reporte de un caso y revisión de la literatura. Rev Med Hosp Gen Mex, 64 (2001), pp. 152-156