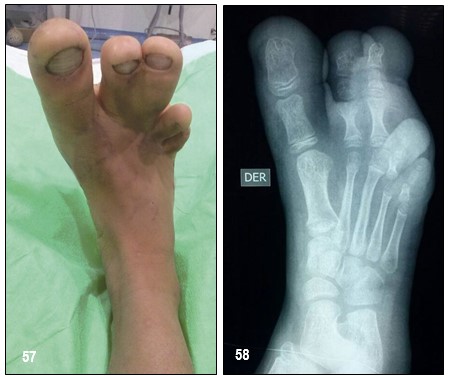
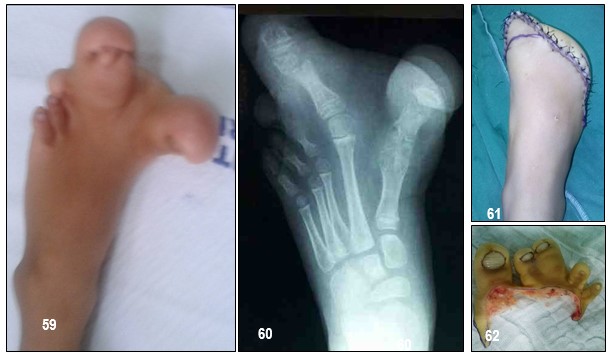
INFORMACIÓN BÁSICA: Encondromatosis. Condroma (ORPHA:296).- Condroma: Es un tumor formador de cartílago, benigno, solitario y de crecimiento muy lento en las metáfisis de los huesos, cuya ubicación más frecuente es central y en las manos. Cuando el tumor es central, se llama encondroma y, cuando es periférico, eucondroma, aunque es mejor hablar de condroma periostal o periférico. Estos últimos son menos frecuentes. Pueden ser lesiones solitarias o múltiples. Las encondromatosis comprenden un grupo heterogéneo de síndromes, difíciles de diferenciar, caracterizados por la presencia de múltiples encondromas que pueden llegar a producir malformaciones musculoesqueléticas (secundarias al acortamiento de extremidades), escoliosis, fracturas patológicas o seudoartrosis. La complicación más temida, el osteocondrosarcoma, puede acontecer hasta en el 25% de los pacientes.
Cuando los condromas o encondromas se transforman en lesiones múltiples se habla de Encondromatosis Familiar Múltiple, que compromete varios huesos y que es motivo de la presentación de uno de los casos que se presentan en tres miembros de una familia. Se habla de Enfermedad de Ollier cuando una encondromatosis múltiple compromete solamente un hemicuerpo, es decir, una encondromatosis múltiple pero unilateral. La mayoría de los autores aceptan esta definición, aunque hay casos de encondromatosis que no corresponden a Enfermedad de Ollier. Cuando esta enfermedad se asocia a hemangiomas de tejidos blandos o flebolitos, se denomina Síndrome de Maffucci. Las imágenes de la 1 a la 6 corresponde a la Enfermedad de Ollier, de la 7 a la a la Encondromatosis Familiar Múltiple. En ambos casos se pueden ver las características típicas de las lesiones.
Las encondromatosis son un grupo de patologías caracterizadas por la presencia de lesiones discondrodisplásicas localizadas en las metáfisis y diáfisis de los huesos en crecimiento, principalmente de las extremidades, no obstante también se han reportado en sitios menos habituales como son: pelvis, costillas, base del cráneo, septum nasal, senos paranasales y tráquea. Dichas lesiones son generalmente benignas, sin embargo su principal complicación es la transformación maligna en condrosarcomas.
La supervivencia a 10 años de los pacientes con condrosarcomas de grado histológico I es hasta del 80%, sin embargo, esta sobrevida disminuye hasta un 31% en tumores de grado histológico III.
La cirugía es la piedra angular del tratamiento debido a la resistencia de los encondromas a la radio y quimioterapia. Reportamos el caso de un paciente con Enfermedad de Ollier, y otros tres con Encondromatosis Múltiple Familiar (Véase índice de enfermedades). A continuación describiremos en detalles ambas entidades. En todo caso, la Tabla 8 (Véase Encondromatosis Múltiple Familiar) se presenta el diagnóstico diferencial de todas las encondromatosis.
INFORMACIÓN BÁSICA.- Enfermedad de Ollier (ORPHA:296/OMIM: /CIE-10: Q78.4) La Enfermedad de Ollier (EO), también conocida como encondromatosis de Ollier, discondrodisplasia, condromatosis interna, encondrosis o condromatosis múltiple asimétrica o sea unilateral, es una afección incluida en el grupo de las ostoecondrodisplasias.
Se caracteriza por lesiones cartilaginosas múltiples dentro de las regiones metafisarias de los huesos largos, tubulares, que se distribuyen de manera bilateral y asimétrica.
Las lesiones parecen surgir de la fisis y ser consecuencia del fallo de osificación encondral normal.
Si los encondromas solitarios representan un 10% de los tumores óseos benignos, la condromatosis múltiple tipo Ollier es 10 veces menos frecuente.
Entidad de aparición precoz, se suele llegar al diagnóstico al detectar tumefacciones o por las deformidades óseas, el dolor local que producen o las fracturas patológicas.
La EO se manifiesta desde la infancia con deformidad de extremidades y secuelas importantes, como alteración del crecimiento, acortamiento con asimetría, fusión articular, sinostosis, etc. La EO es una discondroplasia de osificación encondral descrita originalmente por Ollier en 1900.
Etiología.- Es una enfermedad de etiología desconocida con una baja prevalencia 1/100.000 y presenta una distribución unilateral en muchos casos, por lo que distintos autores han buscado asociaciones genéticas.
Epidemiología.– La incidencia de la EO es baja. Es también posible, que sea una enfermedad infradiagnosticada. La aparición de un encondroma o un condrosarcoma debería llevarnos a descartar la posibilidad de encontrarnos ante una EO o encondromatosis múltiple. Para ello, el método más útil de diagnóstico es la gammagrafía, puesto que las lesiones cartilaginosas suelen ser hipercaptantes con este método de imagen.
Aunque el síndrome de Maffucci y Ollier se asocia con relativa frecuencia a hepatocarcinoma, cáncer de páncreas, tumores de ovario y gliomas, en nuestros pacientes por el momento no hay estigmas de esta asociación.
Ante la escasa incidencia de la enfermedad de Ollier en la población, resulta muy difícil estimar el riesgo de sufrir transformación maligna de los encondromas típicos del síndrome. Ya en 1935 Hunter y Wiles presentaron una revisión de 40 pacientes sin poder determinar el riesgo de malignización. Jeffe en 1958 estima un riesgo de más del 50% de transformación maligna, aunque su serie se basa sólo en el diagnóstico histológico que no es muy fiable para diferenciar encondromas de condrosarcomas de bajo grado.
En el año 2000 Bovée describe un riesgo de 20-50%, aunque su estudio incluyó la necropsia de un paciente afecto de encondromatosis múltiple que falleció por diseminación metastásica pulmonar. Schaison presenta una serie de ocho casos de Ollier con dos transformaciones malignas situadas en fémur. Schwartz presenta una serie de 37 pacientes en la que sólo cuatro se transforman a condrosarcomas.
Herencia.- No se conoce el patrón hereditario de este trastorno, y todos los casos descritos parecen ser esporádicos.
Manifestaciones clínicas.- Desde el punto de vista clínico, suele ser una enfermedad asintomática, sin embargo, en ocasiones provoca fracturas patológicas, deformidades en el crecimiento y dismetrías en la infancia. Al igual que otras enfermedades poliostóticas, existe el riesgo de malignización de alguna de las lesiones hacia condrosarcoma.
En la edad adulta, la presencia de dolor o aumento de tamaño debe hacer sospechar malignización. En ocasiones se asocian a malformaciones vasculares, denominándose en ese caso “síndrome de Maffucci” y a otros tumores como el de la granulosa juvenil.
En otras ocasiones la clínica presentada por los pacientes es una cojera o una asimetría de extremidades.
Sin embargo, un encondroma aislado puede ser un hallazgo casual en una radiografía solicitada por otro motivo. Tal descubrimiento obliga a buscar lesiones en otras localizaciones y, por tanto, a realizar una serie ósea completa.
Los huesos más afectados, después de los de las manos, son los huesos tubulares del pie, el fémur, la tibia, la pelvis, y los huesos del antebrazo.
Características radiológicas.- El aspecto radiológico típico muestra zonas de osteólisis procedentes del cartílago de crecimiento, que se extienden hacia la diáfisis. Se pueden observar calcificaciones en copos de avena en un 50% de los casos.
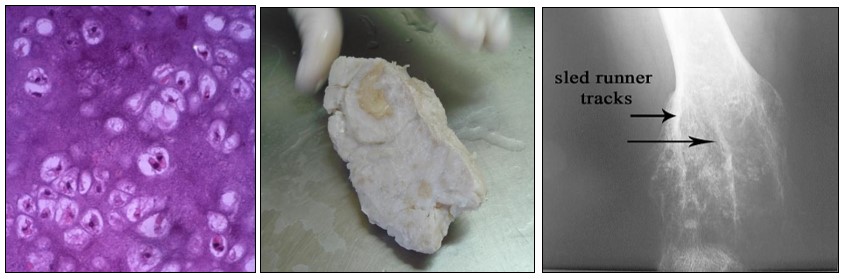
Histología.- Histológicamente la condromatosis múltiple o EO se diferencia del condroma solitario por presentar un tejido más rico en células, con núcleos más grandes (véase imagen con fines didácticos).
Si el diagnóstico de encondroma es evidente, el tratamiento de elección es la observación. La exploración clínica y radiológica deberá pedirse cada 3 meses durante un año, y luego anualmente o cuando los síntomas lo aconsejen. Si el diagnóstico es dudoso, se recomienda curetaje minucioso y el aporte de injerto óseo.
El crecimiento rápido de la lesión o el aumento de dolor aconsejan la evaluación y tratamiento precoz. El método de elección es entonces la resección tumoral asociada o no a un injerto u osteosíntesis.
Diagnóstico.-Radiológicamente se observan múltiples lesiones líticas de aspecto benigno que suelen asentar en las metáfisis aunque tras el cierre fisario pueden extenderse a las epífisis de los huesos largos.
La EO puede ser diagnosticada en forma incidental en un estudio radiológico. La compresión progresiva que producen sobre la corteza de los huesos causa tumefacción y posteriormente deformidad de las superficies afectadas. Los huesos largos, a diferencia de los cortos, presentan menos deformaciones, pero el compromiso prematuro en el crecimiento epifisiario puede producir asimetría y discrepancia en la angulación y longitud de la extremidad. También pueden presentarse dolor y fracturas patológicas por invasión y adelgazamiento de la corteza ósea.
Histológicamente presentan células cartilaginosas más alargadas y binucleadas, similares al condrosarcoma de grado I, por lo que el diagnóstico diferencial resulta difícil y necesita apoyarse en la clínica y en la radiología.
Tratamiento.- Se realiza algún tipo de cirugía para las lesiones benignas; en concreto, legrado y relleno con alo o autoinjerto en las lesiones benignas con o sin osteosíntesis.
Pronóstico.- El diagnóstico precoz de condrosarcoma es fundamental para aumentar la supervivencia de estos pacientes. Las posibilidades de supervivencia disminuyen con la desdiferenciación celular. La alternativa quirúrgica es la base del tratamiento cuando el condrosarcoma es de bajo grado y no presenta afectación metastásica. Es un tumor que no responde a otras modalidades de tratamiento como la quimioterapia o radioterapia. La resección amplia de la lesión, con amputación de los dedos (con reconstrucción) suele ser curativa, procedimiento realizado en esta paciente.
En todo caso el pronóstico dependerá de las complicaciones ortopédicas (asimetría de extremidades, deformidades, fracturas patológicas) y de la malignización de las lesiones.
La asimetría se presenta en un 70% de los pacientes y representa un problema ortopédico importante. Las principales deformidades observadas en esta enfermedad son:
- Ensanchamiento de las falanges,
- Acortamiento asimétrico de las extremidades,
- Encorvamiento de huesos largos y
- Desviación cubital de la muñeca.
Un tercio de los pacientes presenta fracturas patológicas, no sólo en las zonas de condromatosis, sino también a nivel de hueso sano.
El riesgo de desarrollar condrosarcomas secundarios oscila entre un 25 y un 50%. Sin embargo, el 100% de los pacientes afectados de síndrome de Maffucci presentarán degeneración sarcomatosa.
Diagnóstico diferencial.- El diagnóstico diferencial debe incluir la displasia fibrosa, los infartos óseos y el condrosarcoma de bajo grado. La tabla 3 resume el diagnóstico diferencial de las encondromatosis.
Si el diagnóstico de encondroma es evidente, el tratamiento de elección es la observación. La exploración clínica y radiológica debe repetirse cada 3 meses durante un año, y luego anualmente o cuando los síntomas lo aconsejen. Si el diagnóstico es dudoso, se recomienda curetaje minucioso y el aporte de injerto óseo.
El crecimiento rápido de la lesión o el aumento de dolor aconsejan la evaluación y tratamiento precoz. El método de elección es entonces la resección tumoral asociada o no a un injerto u osteosíntesis.
Conclusiones.- Nuestro primer caso cumple con los criterios patognomónicos de una EO descritos por Hunter y Wiles en 1935 y que son:
- 1. Inicio de las manifestaciones clínicas a temprana edad.
- Radiológicamente, cambios limitados a los extremos de los huesos tubulares. Áreas de rarefacción, compromiso secundario de las epífisis. Con imágenes en forma de granos en las metáfisis afectadas y en las epífisis como procesos de crecimiento.
- Tejidos blandos que corresponden a imágenes radiológicas de rarefacción que, cuando son resecadas, corresponden a cartílago.
Bibliografía
Enfermedad de Ollier
- Ollier M. De la dyschondroplasia. Bull Soc Chir Lyon 1899; 3: 22-23.
- H.S. Schwartz, N.B. Zimmerman, M.A. Simon. The malignant potential of enchondromatosis J Bone Joint Surg Am., 69 (1987), pp. 269-274.
- Orphanet: Encondromatosis .
- Jaffe HL; Tumors and tumorous conditions of the bones and joints. Philadelphia: Lea and Febiger, 1968.
- Bovée J; Multiple osteochondromas. Orphanet Journal of Rare Diseases 2008;3:1-7.
- Hunter D, Wiles P. Dischondroplasia (Ollier’s disease). Br J Surg 1935; 22: 507-519.Kumar, V.K. Jain,M. Bharadwaj,R.K. Arya. Ollier disease: Pathogenesis, diagnosis, and management. Orthopedics, 38 (2015), pp. e497-e506
- G.A. Lozano Martínez, J. Llauger Rosselló. Secondary chondrosarcoma: Radiopathological correlation. Radiologia, 57 (2015), pp. 344-359
- Mikola, Z. Yang,K. Merkel,L.A. Gilula. A 7-year-old girl with a growth disturbance in the extremities. Am J Orthop (Belle Mead NJ), 24 (1995), pp. 360-363
- Miyawaki T, Kinoshita Y, Lisuka T. A case of Ollier’s disease of the hand. Ann Plast Surg 1997; 38: 77-80.
- Feldman Frieda. Primary tumors of the hand and carpus. Hand Clinics 1987; 3: 269-289.
- Chew DK, Menelaus MB, Richardson MD. Ollier’s disease: Varus angulation at the lower femur and its management. J Pediatr Orthop 1998; 18: 202-208.
- Hyde GE, Yarington CT Jr, Chu FW. Head and neck manifestations of Maffucci’s syndrome: Chondrosarcoma of the nasal septum. Am J Otolaryngol 1995; 16: 272-275.
- Balcer LJ, Galetta SL, Comblath WT, Liu GT. Neuro-ophthalmologic manifestations of Maffucci’s syndrome ant Ollier’s disease. J Neuro-Ophthalmologic 1999; 19: 62-66.
- Gell JS, Swmrd MW, Ramnani DM, Bradshaw KD. Juvenile granuloma cell tumor in a 13 year old girl with enchondromatosis (Ollier’s disease): A case report. J Pediatr Adolesc Gynecol 1998; 1: 147-150. Ollier M. De la dyschondroplasia. Bull Soc Chir Lyon 1899; 3: 22-23.
- Robbins. Patología estructural y funcional. 6a ed. México: Interamerica, 1990.
- Gabos PG, Bowen JR. Epiphyseal-metaphyseal enchondromatosis. A new clínicasl entity. J Bone Joint Surg Am 1988; 80: 782-792.
- Chan SK, Ng SK, Cho AM, Oh TE. Anaesthetic implications of Maffucci’s syndrome. An Intensive Care 1988; 26: 586-589.
- Scarborough MT, Moreau G. Orthop Clin North Am 1996; 27: 583-589.
- Yahagi H, Osachi S, Toriyama S. Surgical treatment for chondromatosis in childhood. J Jpn Soc Surg Hand 1988; 5: 567-570. Hunter D, Wiles P. Dischondroplasia (Ollier’s disease). Br J Surg 1935; 22: 507-519. 1
- Ozisik YY, Meloni AM, Spanier SS, Busch CH, Kingsley KL, Sandberg AA. Deletion lp in a low-grade chondrosarcoma in a patient with Ollier’s disease. Cancer Genet Cytogenet 1998; 105: 128-133.