INFORMACIÓN BÁSICA: Síndrome de Loeys-Dietz (SLD)(OMIM:609192-610168/ORPHA:60030/CIE-10: Q87.4).- Es un trastorno genético del tejido conjuntivo poco frecuente caracterizado por un amplio espectro de manifestaciones craneofaciales, vasculares y esqueléticas. Se han descrito cuatro subtipos genéticos que forman un continuo clínico. Es una enfermedad autosómica dominante del tejido conectivo, provocada por cambios en los genes TGFBR1, TGFBR2, SMAD3 y TGFB2. Se caracteriza típicamente por aneurismas y tortuosidad arterial, hipertelorismo y úvula bífida o paladar hendido. Lo debemos sospechar ante un paciente con fenotipo marfanoide con las características anteriores.
Historia.- El SLD es un trastorno genético que afecta el tejido conectivo en el cuerpo. El trastorno fue observado y descrito por primera vez por el Dr. Bart Loeys y el Dr. Hal Dietz en la Escuela de Medicina de la Johns Hopkins University en 2005. Desde entonces, otros grupos en todo el mundo han descrito causas genéticas adicionales del síndrome de Loeys-Dietz.
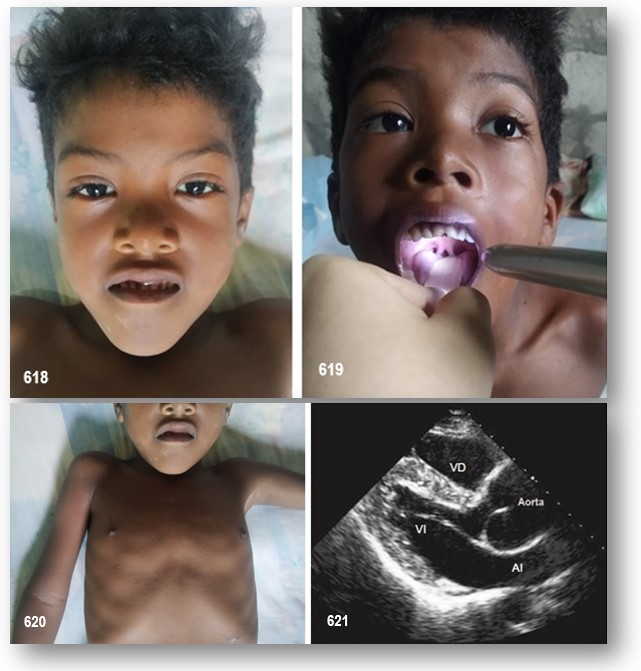
Presentamos el caso de un paciente de 10 años de edad, sin antecedentes familiares de interés, con fenotipo dismórfico, impreso por hipertelorismo, prognatismo mandibular inferior, cráneo turricefálico por craneosinostosis prematura de las suturas coronal y lambdoidea, hendidura palatina e hiperlaxitud articular, úvula bífida y pectus excavatum. Al realizar estudio cardiológico, se evidenció dilatación de la raíz aórtica (25mm, Z-score +4) (Imagen 4). Por cuestiones económicas y logísticas no se pudo llevar a cabo el estudio genético que confirmaría mutación en el gen TGFBR2. Por igual motivo no se puedo ampliar el estudio cardiovascular mediante angio-resonancia magnética.
Características clínicas.- El SLD se caracteriza por hallazgos vasculares (aneurismas y/o disecciones arteriales cerebrales, torácicas y abdominales), manifestaciones esqueléticas (pectus excavatum o pectus carinatum, escoliosis, laxitud articular, aracnodactilia, talipe equinovaro, malformación de la columna cervical y/o inestabilidad), rasgos craneofaciales (ojos muy espaciados, estrabismo, úvula bífida/paladar hendido y craneosinostosis que pueden afectar cualquier sutura) y hallazgos cutáneos (piel aterciopelada y translúcida, fácil formación de moretones y cicatrices distróficas). Las personas con LDS están predispuestas a aneurismas arteriales generalizados y agresivos y complicaciones relacionadas con el embarazo, incluida la rotura uterina y la muerte. Las personas con LDS pueden mostrar una fuerte predisposición a las enfermedades alérgicas/inflamatorias, como asma, eccema, y reacciones a alérgenos alimentarios o ambientales. También hay una mayor incidencia de inflamación gastrointestinal que incluye esofagitis eosinofílica y gastritis o enfermedad inflamatoria intestinal. Se puede ver una gran variación en cómo se distribuyen y se manifiestan los síntomas en personas con LDS, incluso entre miembros de una misma familia que tienen la misma mutación genética.
Diagnóstico.- Si bien varias presentaciones clínicas se han etiquetado en el pasado como LDS tipo I (características craneofaciales presentes), LDS tipo II (características craneofaciales mínimas o ausentes) y LDS tipo III (presencia de osteoartritis) (ver correlaciones de fenotipo por gen), es ahora se reconoce que el LDS causado por una variante patogénica heterocigótica en cualquiera de los seis genes conocidos (ver tabla 1) es un continuo en el que los individuos afectados pueden tener varias combinaciones de características clínicas.
Hallazgos sugerentes.- El SLD debe sospecharse en individuos con los siguientes hallazgos vasculares, esqueléticos, craneofaciales, cutáneos, alérgicos/inflamatorios y oculares [Loeys et al 2005].
Vascular
- Dilatación o disección de la aorta y otras arterias. La dilatación de la raíz aórtica se observa en más del 95% de los probandos; la raíz aórtica es el sitio más común de disección. En raras ocasiones, se pueden observar aneurismas o disecciones en otras arterias de la cabeza, el tórax, el abdomen o las extremidades en ausencia de afectación aórtica.
- Otros aneurismas arteriales y tortuosidad
- La evaluación se realiza mejor con angiografía por resonancia magnética o angiografía por tomografía computarizada con reconstrucción 3D de la cabeza a la pelvis para identificar aneurismas o disecciones arteriales y tortuosidad arterial en todo el árbol arterial. La tortuosidad suele ser más prominente en los vasos de la cabeza y el cuello.
- Aproximadamente el 50% de los individuos con SLD estudiados tenían un aneurisma distante de la raíz aórtica que no habría sido detectado por ecocardiografía.
Esquelético
- – Pectus excavatum o pectus carinatum
- – Escoliosis
- – Laxitud o contractura de las articulaciones (que generalmente involucra los dedos)
- – Aracnodactilia
- – Talipes equinovarus
- – Malformación y / o inestabilidad de la columna cervical
- – Osteoartritis
- – Craneofacial
- – Ojos muy espaciados
- – Úvula bífida/paladar hendido
- – Craneosinostosis, en la que puede estar involucrada cualquier sutura
Cutáneo
- – Piel suave y aterciopelada
- – Piel translúcida con venas subyacentes fácilmente visibles
- – Moretones con facilidad
- – Cicatrices distróficas
- – Milia, prominente en la cara.
Enfermedad alérgica / inflamatoria
- Alergias a los alimentos
- Alergias estacionales
- Asma/sinusitis crónica
- Eczema
- Esofagitis/gastritis eosinofílica
- Enfermedad inflamatoria intestinal
Ocular
- – Escleróticas azules u oscuras
- – Establecimiento del diagnóstico
Características sistémicas compatibles que incluyen manifestaciones craneofaciales, esqueléticas, cutáneas y/o vasculares características que se encuentran en combinación. Se da especial énfasis a la tortuosidad arterial, que incluye de manera prominente los vasos de la cabeza y el cuello, y a los aneurismas o disecciones que involucran arterias musculares medianas a grandes en todo el árbol arterial.
Nota: Si hay antecedentes familiares de SLD documentado, se puede diagnosticar a los familiares en riesgo mediante pruebas genéticas moleculares, incluso si la afectación vascular u otras características aún no son evidentes.
Diagnóstico/molecular.- El diagnóstico de SLD se establece en un probando (por definición, una persona sin antecedentes familiares conocidos de SLD) que tiene una variante patogénica heterocigótica en SMAD2, SMAD3, TGFB2, TGFB3, TGFBR1 o TGFBR2 (ver Tabl 1) y o bien, de los siguientes [MacCarrick et al 2014]:
- – Agrandamiento de la raíz aórtica (definido como una puntuación z de la raíz aórtica ≥2.0) o disección tipo A.
- – Características sistémicas compatibles que incluyen manifestaciones craneofaciales, esqueléticas, cutáneas y/o vasculares características que se encuentran en combinación. Se da especial énfasis a la tortuosidad arterial, que incluye de manera prominente los vasos de la cabeza y el cuello, y a los aneurismas o disecciones que involucran arterias musculares medianas a extensas en todo el árbol arterial.
Nota: En presencia de antecedentes familiares de LDS documentado, el diagnóstico se puede realizar en familiares en riesgo sobre la base de pruebas genéticas moleculares, incluso si la afectación vascular u otras características aún no son evidentes (consulte evaluación de familiares en riesgo).
El diagnóstico de SLD se establece en individuos con base en hallazgos clínicos característicos en el probando y miembros de la familia y/o por la identificación de una variante patogénica heterocigota en SMAD2, SMAD3, TGFB2, TGFB3, TGFBR o TGFBR2.
Correlaciones de fenotipos por gen.– Varias presentaciones clínicas se han etiquetado en el pasado como SLD tipo I (características craneofaciales presentes), SLD tipo II (características craneofaciales mínimas o ausentes), SLD tipo III (presencia de osteoartritis), etc. Estas designaciones de subtipos proporcionan una indicación general del espectro de gravedad de la enfermedad, desde la más a la menos grave: LDS1 = LDS2> LDS3> LDS4> LDS5.
Nota: Todavía no hay suficiente información sobre el espectro de características de SLD causadas por variantes patogénicas heterocigotas en SMAD2 para colocar este gen en el continuo o en la Tabla 50.

Los enfoques de las pruebas genéticas moleculares pueden incluir una combinación de pruebas dirigidas a genes (pruebas seriadas de un solo gen o un panel multigénico) y pruebas genómicas (secuenciación genómica completa) según el fenotipo.
Las pruebas dirigidas a genes requieren que el médico determine qué genes probablemente estén involucrados, mientras que las pruebas genómicas no lo hacen. Debido a la superposición clínica, es difícil predecir cuál de los genes conocidos relacionados con SLD será el causante en cualquier individuo afectado. Aunque es probable que los individuos con los hallazgos distintivos de SLD descritos sean diagnosticados mediante pruebas dirigidas a genes, aquellos que no tienen características de discriminación suficiente para considerar el diagnóstico de SLD tienen más probabilidades de ser diagnosticados mediante pruebas genómicas.
Diagnóstico diferencial.- Con formas sindrómicas de aneurismas de la aorta torácica:
El síndrome de Marfan es un trastorno sistémico con un alto grado de variabilidad clínica. Las manifestaciones cardinales involucran los sistemas ocular, esquelético y cardiovascular [Judge & Dietz 2005]. Las manifestaciones cardiovasculares incluyen dilatación de la aorta en los senos de Valsalva, predisposición a rotura y desgarro aórtico, prolapso de la válvula mitral con o sin regurgitación, prolapso de la válvula tricúspide y agrandamiento de la arteria pulmonar proximal. El síndrome de Marfan es causado por una mutación de FBN1 y se hereda de forma autosómica dominante.
El síndrome de Shprintzen-Goldberg (SGS) se caracteriza por craneosinostosis, características craneofaciales distintivas, cambios esqueléticos, anomalías neurológicas, discapacidad intelectual leve a moderada y anomalías cerebrales. Pueden ocurrir anomalías cardiovasculares (prolapso de la válvula mitral, regurgitación mitral e insuficiencia aórtica), pero la dilatación de la raíz aórtica se observa con menos frecuencia que en el LDS y puede ser leve. Una característica importante que distingue SGS de LDS es la incidencia casi uniforme de retraso en el desarrollo en SGS.
El análisis molecular de una serie de individuos con SGS típico no reveló variantes patogénicas en TGFBR1 o TGFBR2 [Loeys et al 2005]. Los individuos afectados suelen ser casos simplex (es decir, sin antecedentes familiares de SGS), aunque se han descrito casos raros de herencia autosómica dominante aparente.
La mayoría de los individuos con SGS tienen una variante patógena de novo heterocigota sin sentido en SKI [Carmignac et al 2012, Doyle et al 2012]. La proteína SKI es un conocido inhibidor de la señalización de TGFβ, que conecta funcionalmente SGS con LDS.
Nota: Un individuo reportado con SGS por Kosaki et al [2006] que considero tenían SLD basado en la presencia de tortuosidad arterial y una úvula bífida [Robinson et al 2006].
El síndrome de aneurisma aórtico asociado a BGN es una afección ligada al cromosoma X causada por una variante patógena hemicigótica en BGN, que codifica un proteoglicano llamado biglicano [Meester et al 2017a]. Las características clínicas son muy similares a las del síndrome de Marfan y el síndrome de Loeys-Dietz. Esto incluye la dilatación y disección temprana de la raíz aórtica, ojos muy separados, hipermovilidad en las articulaciones, contracturas, úvula bífida y deformidades en el pecho.
En algunas familias, las hembras heterocigotas también se ven afectadas. El tipo de variantes patogénicas en BGN sugiere pérdida de función como mecanismo de enfermedad. MASS fenotipo (OMIM 604308) se caracteriza por válvula mitral de prolapso, o borderline y no progresiva, una ampliación aortica, y no específica cutáneo y esqueléticas, hallazgos que se superponen con las que se observan en el síndrome de Marfan.
Uno tiene más confianza en este diagnóstico cuando se observan manifestaciones concordantes en varias generaciones de una familia determinada. Sin embargo, algunos individuos de dicha familia podrían estar predispuestos a una afectación vascular más grave y, por lo tanto, debe mantenerse un régimen de imágenes cardiovasculares intermitentes. Es difícil distinguir el fenotipo MASS del síndrome de Marfan «emergente» al evaluar un caso (es decir, ocurrencia única en una familia), especialmente durante la niñez. Las variantes heterocigotas en FBN1 pueden ser la causa. La herencia es autosómica dominante.
Los síndromes de Ehlers-Danlos
La aracnodactilia contractural congénita (CCA) se caracteriza por una apariencia similar a la de Marfan (habitus alto y delgado en el que la extensión del brazo excede la altura) y dedos de manos y pies largos y delgados (aracnodactilia). Se ha informado un agrandamiento progresivo de la aorta ascendente en los senos de Valsalva, pero no hay evidencia de que la dilatación aórtica progrese a disección o ruptura [Gupta et al 2002]. Se han visto bebés con una forma grave que presenta muchas anomalías en el corazón y el sistema digestivo, además de los problemas esqueléticos típicos. La CCA está causada por una mutación de FBN2 y se hereda de forma autosómica dominante.
El síndrome de tortuosidad arterial (ATS) es un trastorno del tejido conectivo autosómico recesivo poco común, caracterizado principalmente por tortuosidad severa, estenosis y aneurismas de la aorta y arterias de tamaño mediano [Wessels et al 2004]. También es común la afectación esquelética y cutánea. El defecto genético subyacente es la homocigosidad para las variantes de pérdida de función en SLC2A10, el gen que codifica la familia de transportadores de solutos 2, miembro transportador de glucosa facilitado 10. Aunque no se esperaría que un defecto del transportador de glucosa cause un patrón arterial anormal, estudios adicionales indicaron una la vía de señalización de TGFβ [Coucke et al 2006], consistente con la fisiopatología en SLD y síndrome de Marfan.
Asesoramiento genético.– El SLD se hereda de forma autosómica dominante. Aproximadamente el 25% de las personas diagnosticadas con SLD tienen un padre afectado; aproximadamente el 75% de los probandos tienen SLD como resultado de una variante patogénica de novo. Cada hijo de un individuo con SLD tiene un 50% de posibilidades de heredar la variante patógena y el trastorno. El diagnóstico prenatal de embarazos con mayor riesgo de SLD es posible si se conoce la variante patogénica en la familia.
Tratamiento.- La afectación cardiovascular en el SLD es muy frecuente. Los pacientes afectados tienen alto riesgo de disección o rotura aórtica a edades muy tempranas, incluso con diámetros aórticos no muy dilatados. Se recomienda iniciar tratamiento con betabloqueante o con antagonistas de los receptores de la angiotensina, además de realizar estudios periódicos de imagen que incluyan la aorta y sus ramas. Una de las dificultades para detectar la enfermedad es que se confunde con el síndrome de Marfan, pero con peor pronóstico, ya que los pacientes fallecen a edades más tempranas. El diagnóstico precoz es fundamental, ya que un tratamiento quirúrgico adecuado podría disminuir las complicaciones.
Bibliografia
- MacCarrick, J.H. Black, S. Bowdin, I. El-Hamamsy, P.A. Frischmeyer-Guerrerio, A.L. Guerrerio, et al. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet Med, 16 (2014), pp. 576-587 http://dx.doi.org/10.1038/gim.2014.11| Medline E. Fortuny, V. Cañadas, I. Vilacost.
- Aortic aneurysm in hereditary syndromes. Differential diagnosis of Marfan syndrome. Cardiocore, 46 (2011), pp. 105-108
- J.A. Williams, J.M. Hanna, A.A. Shah, N.D. Andersen, M.T. McDonald, Y.H. Jiang, et al. Adult surgical experience with Loeys-Dietz syndrome. Ann Thorac Surg, 99 (2015), pp. 1275-1281 http://dx.doi.org/10.1016/j.athoracsur.2014.11.021 | Medline
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Síndromes de aneurismas causados por mutaciones en el receptor TGF-beta. N Engl J Med. 2006; 355 : 788–98. [ PubMed ]
- Wessels MW, Catsman-Berrevoets CE, Mancini GM, Breuning MH, Hoogeboom JJ, Stroink H, Frohn-Mulder I, Coucke PJ, Paepe AD, Niermeijer MF, Willems PJ. Tres nuevas familias con síndrome de tortuosidad arterial. Am J Med Genet A. 2004; 131 : 134–43. [ PubMed ].
- Robinson P, Neumann L, Tinschert S. Respuesta a Kosaki et al, Patología molecular de Shprintzen-Goldberg. Am J Med Genet A. 2006; 140 : 109-10. [ PubMed ]
- Carmignac V, Thevenon J, Adès L, Callewaert B, Julia S, Thauvin-Robinet C, Gueneau L, Courcet JB, Lopez E, Holman K, Renard M, Plauchu H, Plessis G, De Backer J, Child A, Arno G , Duplomb L, Callier P, Aral B, Vabres P, Gigot N, Arbustini E, Grasso M, Robinson PN, Goizet C, Baumann C, Di Rocco M, Sanchez Del Pozo J, Huet F, Jondeau G, Collod-Beroud G , Beroud C, Amiel J, Cormier-Daire V, Rivière JB, Boileau C, De Paepe A, Faivre L.Las mutaciones en marco en el exón 1 de SKI causan el síndrome de Shprintzen-Goldberg dominante. Soy J Hum Genet. 2012; 91 : 950–7
- Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A. Las mutaciones en el transportador facilitador de glucosa GLUT10 alteran la angiogénesis y causan el síndrome de tortuosidad arterial. Nat Genet. 2006; 38 : 452–7
- Gupta PA, Putnam EA, Carmical SG, Kaitila I, Steinmann B, Child A, Danesino C, Metcalfe K, Berry SA, Chen E, Delorme CV, Thong MK, Ades LC, Milewicz DM. Diez mutaciones novedosas de FBN2 en aracnodactilia contractural congénita: descripción de la patogenia molecular y el fenotipo clínico. Hum Mutat. 2002; 19 : 39–48.
- Kosaki K, Takahashi D, Udaka T, Kosaki R, Matsumoto M, Ibe S, Isobe T, Tanaka Y, Takahashi T. Patología molecular de Shprintzen-Goldberg. Am J Med Genet A. 2006; 140 : 104–8.
- van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collée M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM. Las mutaciones en SMAD3 causan una forma sindrómica de aneurismas aórticos y disecciones con osteoartritis de inicio temprano. Nat Genet. 2011; 43 : 121–6.
- Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB. Proyecto de secuenciación Go Exome del Instituto Nacional del Corazón, los Pulmones y la Sangre (NHLBI), Jondeau G, Milewicz DM. Las mutaciones de TGFB2 causan aneurismas de la aorta torácica familiar y disecciones asociadas con características sistémicas leves del síndrome de Marfan. Nat Genet. 2012; 44 : 916-21.
- Judge DP, Dietz HC. Síndrome de Marfan. Lanceta. 2005; 366 : 1965–76.
- Lindsay ME, Dietz H. Lecciones sobre la patogenia del aneurisma de enfermedades hereditarias. Naturaleza. 2011; 473 : 308–16.
- Wischmeijer A, Van Laer L, Tortora G, Bolar NA, Van Camp G, Fransen E, Peeters N, di Bartolomeo R, Pacini D, Gargiulo G, Turci S, Bonvicini M, Mariucci E, Lovato L, Brusori S, Ritelli M , Colombi M, Garavelli L, Seri M, Loeys BL. Aneurisma de la aorta torácica en la infancia en el síndrome de aneurisma-osteoartritis debido a una nueva mutación SMAD3: descripción adicional del fenotipo. Am J Med Genet A. 2013; 161A : 1028–35.