INFORMACIÓN BÁSICA.- SÍNDROMES PTERIGIUM MÚLTIPLES CONGÉNITOS (OMIM 265000/ORPHA:2990).- Síndromes pterigium múltiples (MPS) son un grupo de trastornos caracterizados por anomalías congénitas múltiples, forman redes (pterigión) en el cuello, los codos y/o rodillas y contracturas articulares (artrogriposis). Las MPS son fenotípica y genéticamente heterogéneas, pero se dividen tradicionalmente en 2 tipos: prenatal letales y no letales, este ultimo denominado Síndrome de Escobar o MPS tipo B. Contracturas congénitas características del síndrome de Escobar pueden ser causadas por los movimientos fetales reducidos en períodos sensibles del desarrollo. Las posibles causas de la movilidad fetal disminuida incluyen la miastenia gravis.
El Pterigium múltiple letal es una condición heredada, la cual se caracteriza por la presencia de pterigium articulares, contracturas en flexión, asociadas a otros defectos como retraso del crecimiento intrauterino, hydrops fetal, higroma cístico, artrogriposis y pulmones hipoplásicos; el pterigium múltiple letal es incompatible con la vida extrauterina. Es una forma de Artrogriposis múltiple congénita caracterizada por contracciones congénitas y escoliosis.
El MPS fue inicialmente descrito por Bussiere en 1902. Frawley en 1925, describió las características de este síndrome, y Gorlin y cols en 1976, utilizaron la designación de este para describir una entidad autosómica recesiva. Posteriormente Escobar y cols en 1978, realizan una revisión y descripción detallada del síndrome por lo cual actualmente también se le conoce a una variante menos severa de este como variante Escobar o síndrome Escobar.
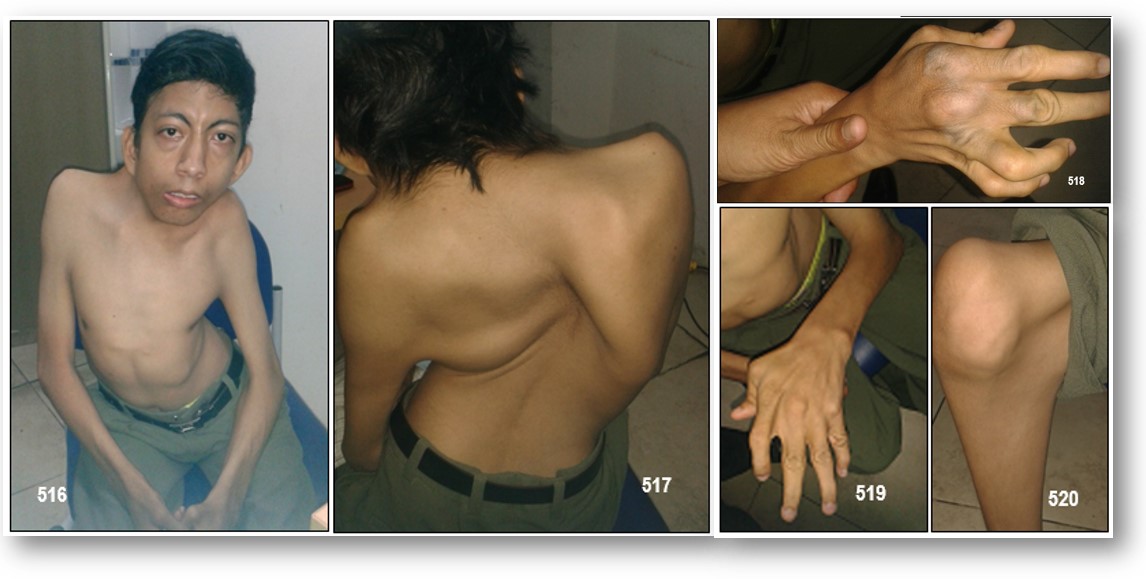
SÍNDROME DE PTERIGIUM MÚLTIPLE NO LETAL O SÍNDROME DE ESCOBAR ( ORPHA: 2990/OMIM:265000-618469/CIE-10:Q79.8 ).-Es un síndrome dismórfico/ con anomalías congénitas múltiples, de origen genético y poco frecuente, caracterizado por pterigium (pliegues) congénito que afecta principalmente al cuello y a las grandes articulaciones, artrogriposis múltiple, talla baja y dismorfia craneofacial (incluyendo ptosis, fisuras palpebrales descendentes, paladar ojival y retrognatia). Otras manifestaciones incluyen disminución de los movimientos, debilidad facial, dificultad respiratoria, anomalías vertebrales, escoliosis, anomalías de los dedos de las manos y criptorquidia, entre otras. La enfermedad es una variante no letal del síndrome de pterigium múltiple.
Haciendo un poco de historia, a principios del siglo pasado, Bussiere y Frawley hicieron las primeras descripciones de este síndrome, al cual le dieron el nombre de Pterygium Múltiple Congénito, ya que desde el nacimiento se pueden apreciar los Pterigion múltiples y las alteraciones musculoesqueléticas, pero fue hasta 1978 que Víctor Escobar y Cols, publicaron un artículo donde él describió más ampliamente las características clínicas de este síndrome. Las alteraciones que describió en estos pacientes son parte de las dismorfias mayores y menores.
El síndrome de Escobar es una entidad poco común y por lo tanto poco conocida, ya que en la bibliografía reciente sólo están reportados 100 casos a nivel mundial; este síndrome tiene como sinónimo Pterygium múltiple congénito.
Una parte importante de este síndrome es la alteración pulmonar restrictiva con la que pueden llegar a cursar, esto debido a debilidad muscular y restricción en la movilidad de la caja torácica más la escoliosis que agrava la misma.
Etiología.- La etiología de esta condición es causada por mutaciones homocigotas o compuestas heterocigotas en el gen CHRNG, locus 2q37, el cual codifica la subunidad gamma del receptor de acetilcolina (AChR). Mutaciones en este gen también han sido asociadas con las variantes no letales de este síndrome.
Manifestaciones clínicas.– El síndrome de Escobar o síndrome del pterigyum múltiple no letal, es un desorden raro caracterizado por dismorfia general, aspecto inexpresivo o falta de emoción, pterygium coli lateral y anterior en cuello, pterigyum en axilas, codos, camptodactilia, sindactilia cutánea y pulgares flexionados, xifoescoliosis, contractura en flexión de rodillas e ingle por pterigyum y pies en mecedora. Y puede estar acompañado en algunas ocasiones con luxación de cadera, ausencia de rótulas, hernias inguinales, platispondilia, espina bífida oculta y criptorquidia con inteligencia normal.

Herencia.– Este síndrome se hereda de forma autosómica recesiva. Aún no está definida la etiología y fisiopatología, se sugiere como un desorden neuromuscular, sin embargo estudios de secuenciación genética han demostrado una mutación puntual.
El síndrome de pterygium múltiple o síndrome Escobar no letal es una enfermedad rara que es una variante de síndromes múltiples de pterigion. Se diagnostica por ecografías uterinas y luego se ratifica en el período neonatal. Un caso de Escobar neonatal no letal se manifestó en un bebé de 35 semanas y 6 días que presentó intraútero disminución del movimiento fetal, oligohidramnios y artrogriposis.
El síndrome de Escobar es una entidad muy rara, a mediados de los 80 había descritos menos de 25 casos; actualmente se han descrito 100 casos a nivel mundial, por tal motivo exponemos este caso clínico.
Genética.- Se han identificado mutaciones causantes de miastenia grave en genes que codifican para diferentes subunidades del receptor de acetilcolina (AchR).
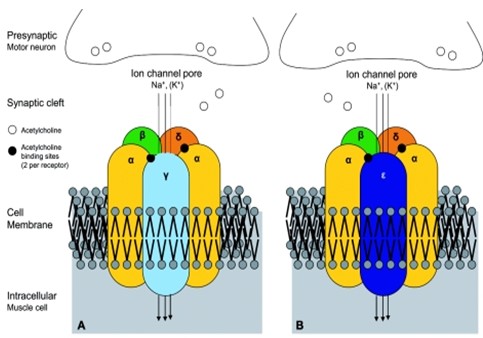
El gen CHRNG, situado en el cromosoma 2 (q23-q34), gen que codifica para la subunidad gamma (fetal) del receptor AchR, expresándose antes de la semana 33 de gestación, y siendo posteriormente desplazada por la subunidad épsilon que dará lugar al receptor AchR adulto. Mutaciones recesivas nonsense y missense en la subunidad gamma (fetal) del receptor de acetilcolina (CHRNG) pueden causar tanto las formas letal como la no letal del síndrome de pterigium múltiple.
Las mutaciones que causan el síndrome miasténico congénito se identificaron en las subunidades de la CADH. CHRNG El gen que codifica la subunidad gamma de la CADH se expresa antes y 33a semanas de gestación en los seres humanos, pero la subunidad épsilon se sustituye por el último período fetal o perinatal, formando de esta manera el adulto AChR. Hubo informes de que las mutaciones recesivas absurdo y sin sentido en la subunidad gamma del receptor de acetilcolina embrionario (CHRNG) pueden causar letal y no letal MPS, lo que demuestra que el pterigión resultado de acinesia fetal.
Diagnóstico.- El diagnostico del Síndrome de Escobar no es sencillo, frecuentemente se diagnostica como artrogriposis, síndrome dismórfico, síndrome dismorfológico, enanismo, entre otros. El diagnóstico resulta de un análisis de la secuencia positiva para el gen CHRNG después del alta. La presentación clínica del síndrome de Escobar no letal, así como las estrategias de diagnóstico y tratamiento posterior se basan en planes de cuidado continuado.
Discusión.- Describimos un paciente con diagnóstico de síndrome de Escobar o pterigium múltiple con manifestaciones clínicas características. Destacamos la importancia del seguimiento clínico del paciente para cuyas características se planteó una etiología genética. El fenotipo clínico completó su definición con la edad; en este sentido, algunos elementos de incipiente manifestación, como el pterigium (pilar fundamental para el diagnóstico), se hizo más evidente -así como otras características faciales- que permitieron, al reevaluarlo a los 6 meses después de la primera consulta, establecer rápidamente el diagnóstico nosológico de una entidad poco frecuente. Para realizar el diagnóstico diferencial con otras formas de pterigium múltiple se deben considerar las siguientes entidades: artrogriposis congénita múltiple, pterigium poplíteo y la presentación de pterigium en forma aislada. (Tabla 1).
La patogenia y fisiopatología del síndrome de Escobar o síndrome del pterigium múltiple no letal aún no está bien definida. Se sugiere un desorden neuromuscular; si bien los estudios de electromiografía y velocidad de conducción resultan normales, los estudios patológicos de biopsia muscular muestran la presencia de degeneración muscular y desorganización de miofibrillas, y los estudios inmunohistoquímicos e inmunofluorescentes no muestran una reacción específica. Según Hoffmann y colaboradores, el síndrome de Escobar podría estar causado por una mutación en el gene CHRNG que codifica la subunidad g del receptor de la acetilcolina (AChR). (Fig. 1) El AChR fetal se expresa en sitios y momentos de la vida fetal que se corresponden con el desarrollo del fenotipo clínico del síndrome.
El receptor de la acetilcolina ayuda a establecer la unión primaria entre el músculo y el axón por lo que la subunidad g no sólo contribuye a la transducción de la señal neuromuscular sino que también es importante para la organogénesis neuromuscular.
Hoffmann et al. (2006) y Morgan et al. (2006) demostraron que tanto la forma letal y los no letales de síndrome de Escobar o síndrome de pterigium múltiple pueden ser causados por mutaciones en la subunidad gamma fetal del receptor de acetilcolina nicotinérgico. Hoffmann et al. (2006) encontraron 8 mutaciones en 7 familias con síndrome de Escobar. Morgan et al. (2006) encontraron 6 mutaciones homocigóticas en 6 familias con variante letal del síndrome de Escobar. En una familia ambas variantes estaban presentes. Hoffmann et al. En ese mismo año, observaron que las contracturas congénitas características del síndrome de Escobar pueden ser causadas por los movimientos fetales reducidos en períodos sensibles de desarrollo. Las posibles causas de la movilidad fetal disminuida incluyen restricciones de espacio como oligohidramnios, drogas, enfermedades metabólicas o trastornos neuromusculares incluyendo miastenia gravis. Miastenia se caracteriza por debilidad muscular intermitente, causada por autoanticuerpos que se unen a receptores de acetilcolina nicotinérgico (CAIS). Durante el embarazo, los anticuerpos pueden atravesar la placenta y causar debilidad muscular transitoria o, algo más serio, como una artrogriposis o el síndrome.
Las mutaciones del gene CHRNG originarían una subunidad g mutada o ausente que impediría la expresión del AChR fetal reduciendo la fuerza y el movimiento muscular prenatal. Esto explicaría consecuencias dismórficas como contracturas, pterigium, paladar alto y criptorquidia en varones.
Dado que la subunidad g generalmente desaparece en el desarrollo fetal tardío siendo sustituida por la subunidad e, los pacientes no presentan signos o avances de miastenia luego del nacimiento, a diferencia de otros pacientes con mutaciones en otras subunidades del AChR. (Figura 38).
Según Vogt J y col 2012 plantea que las mutaciones de línea germinal en el gen CHRNG que codifica la subunidad γ del receptor de acetilcolina embrionaria pueden provocar la variante letal de Escobar (EVMPS) o la forma letal (LMPS) de síndrome pterigium múltiple o (MPS).
El papel del receptor de la acetilcolina γ, subunidad fetal en la transducción de señales neuromuscular γ en el establecimiento del encuentro primario del músculo y el terminal del nervio motor; las mutaciones en el CHRNG descritos en el síndrome de Escobar pueden causar una interrupción más amplio de proteínas postsinápticas y resultar en el desarrollo aberrante de la unión neuromuscular en la transmisión neuromuscular prenatal alterada y/o sinaptogénesis neuromuscular anormal. (Robinson KG y col. 2013).
Es posible que las mutaciones en estos genes se encuentren en la secuencia promotora o en las regiones intrónicas profundas que no se pudo identificar o el síndrome de Escobar es causado por mutaciones de un gen diferente (Kodaganur y col 2013)
Si bien, hasta el momento, no se ha podido establecer el defecto molecular, el diagnóstico clínico permite ofrecer asesoramiento genético respecto del pronóstico y tratamiento del paciente, así como del riesgo de recurrencia de igual patología en la familia, especialmente para la hermana y las dos medio hermanas, clínicamente normales que, por tratarse de una enfermedad genética con mecanismo de herencia autosómico y recesivo de baja frecuencia presentan muy bajo riesgo de descendencia afectada con parejas no consanguíneas.
En relación al síndrome Pterigiun antecubital, este es un trastorno autosómico dominante caracterizado por una red carnosa extiende a través de la cara anterior de la fosa cubital, la ausencia de la porción larga del tríceps, limitación de la extensión del codo completa, y desaparecidos pliegues de la piel sobre las articulaciones interfalángicas terminales de los dedos (Wallis et al 1988).
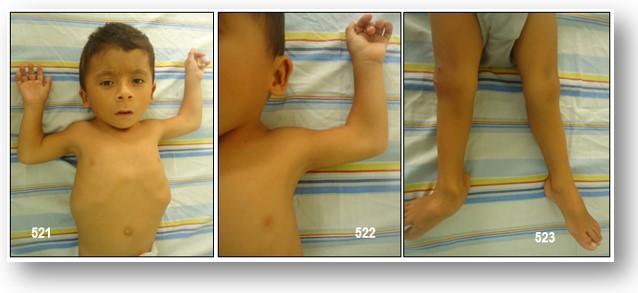
SÍNDROME DE PTERIGIUM MULTIPLE LETAL (ORPHA:33108/OMIM:253290/CIE-10:Q79.8).- El síndrome de Pterigium múltiple letal, es un desorden letal caracterizado por la presencia de múltiples pterigiums, hidrops, higroma cístico, siendo el pterigium la característica obligatoria de este, caracterizado además por un retraso del crecimiento intrauterino, acinesia fetal, contracturas articulares múltiples que resultan en artrogriposis grave y pterigium (pliegues) en múltiples articulaciones. Se entiende como pterigium a los pliegues de piel a lo largo de la articulación, provocando contracturas articulares en flexión y consecuentemente artrogriposis.
También se encuentra asociado a la presencia de otras alteraciones morfológicas frecuentes como son: hipoplasia pulmonar, facies anormales con hipertelorismo, fisuras palpebrales inclinadas hacia abajo, nariz plana, boca pequeña, paladar hendido, micrognatia, orejas de implantación baja, retardo del crecimiento intrauterino, hipoplasia cardiaca, hernia diafragmática, defectos oculares (cataratas), anormalidades renales (hidronefrosis, displasia renal), anormalidades esqueléticas, cordón umbilical corto.
Epidemiología.- Hasta la fecha se han descrito menos de 50 casos de fetos en 28 familias. De éstos, aproximadamente el 60% eran de sexo masculino. La mitad de las familias sólo contaba con varones afectos, incluyendo cinco familias con varios varones.
Etiología.- La fisiopatología de esta afección es desconocida, pero estudios de genética molecular han demostrado que mutaciones causales del síndrome en las subunidades del receptor de acetilcolina codificadas por los genes CHRNA1 (2q31.1), CHRND (2q37.1) y CHRNG (2q37.1), así como en el gen de la nebulina (NEB, 2q23.3) y del receptor de rianodina (RYR1, 19q13.2). No obstante Meyer-Cohen y cols, describen también la posibilidad de herencia ligada al cromosoma X recesivo y está documentado en la literatura casos de fetos abortados de padres primos en primer grado de origen marroquí con diagnóstico de Pterigium múltiple letal.
Se han reportado casos donde existe una relación de consanguinidad, lo cual sugiere una herencia recesiva autosómica y probable mutación en los genes CHRNG, CHRNA1 y CHRND; sin embargo cuando el paciente es de sexo masculino, como el caso que presentamos, no se puede descartar una herencia recesiva ligada al cromosoma X. En este caso y en los que el feto tiene un defecto congénito incompatible con la vida, lo ideal es tomar las muestras de sangre in útero cuando el feto aún está vivo, con lo cual se lograra con mayor probabilidad cultivo celular para cariotipo y extracción de DNA. Teniendo en cuenta las dificultades para realizar pruebas moleculares de defectos congénitos de baja prevalencia como en el Pterigium múltiple letal, el diagnóstico clínico toma trascendental importancia.
Sumando los hallazgos en las ecografías prenatales, las radiografías y autopsia se logra hacer un diagnóstico. Así es que todos estos antecedentes son trascendentales para el análisis del caso por un grupo multidisciplinario integrado por obstetras, especialistas en diagnóstico prenatal o medicina materna fetal, radiólogos, patólogos y genetistas.
Métodos diagnósticos.- El diagnóstico se sospecha en base a los hallazgos clínicos y ecográficos observados en el seguimiento rutinario del embarazo (para más detalles, consulte la sección sobre diagnóstico prenatal).
Diagnóstico diferencial.- El diagnóstico diferencial incluye otros trastornos que se presentan con características ecográficas prenatales de movimiento fetal reducido o ausente asociado a una postura fetal anómala y a otras condiciones artrogripóticas. Estos trastornos pueden incluir la secuencia de deformación por acinesia fetal (SDAF), los síndromes de Bartsocas-Papas y de pterigión múltiple variante de Escobar, la artrogriposis múltiple congénita y la miastenia gravis materna, así como la trisomía 18, defectos graves del tubo neural, la secuencia de regresión caudal y anomalías vertebrales, el complejo extremidad-pared abdominal, masas en el cuello fetal, la hipoxia fetal, el síndrome de anillos de constricción y la constricción fetal.
Diagnóstico prenatal.- La acinesia fetal puede detectarse a las 12 semanas. Los hallazgos ecográficos prenatales de LMPS incluyen retraso del crecimiento intrauterino, contracturas en flexión de las extremidades, pterigium múltiple, higroma quístico, hidropesía fetal, pulmones hipoplásicos, paladar hendido y otras anomalías estructurales. Los hallazgos adicionales de desarrollo esquelético hipoplásico pueden ayudar a distinguir el LMPS de otros trastornos con SDAF, mientras que la ecografía detallada y la RM fetal permiten identificar anomalías del sistema nervioso central que pueden ser un hallazgo del LMPS o sugerir una etiología alternativa. Además, cuando la mutación ha sido previamente identificada, se pueden realizar pruebas genéticas moleculares prenatales si las exploraciones iniciales no son concluyentes. También se debe sospechar LMPS en pacientes con antecedentes de pérdidas recurrentes en el segundo trimestre de gestación.
El diagnóstico prenatal de esta patología es difícil, dada la dificultad en la visualización de los pterigium, sin embargo la presencia de higroma cístico, hidrops y contracturas en las extremidades en flexión marcada y sin movimientos, orientan el diagnóstico. Este empieza idealmente en la ecografías de las 11 a 14 semanas y 20 a 24 semanas, donde se deben identificar los hallazgos y solicitar una ecografía de detalle anatómico o nivel III. Con equipos de alta resolución y personal mejor entrenado se deberá hacer una observación milimétrica de toda la anatomía para configurar el síndrome con las características clásicas y otras frecuentemente asociadas al Pterigium múltiple letal, y así diferenciarlas con las de la artrogriposis múltiple congénita, el síndrome de Pena- Shokeir, la distrofia congénita, y el síndrome de Escobar o Pterigium no letal.
Consejo genético.- El patrón de herencia es autosómico recesivo, con un riesgo de recurrencia entre hermanos del 25%. En algunos casos, se ha descrito la herencia ligada al cromosoma X, como lo sugiere la mayor proporción de varones con LMPS.
Manejo y tratamiento.- Dado que el trastorno es letal, es posible proponer la interrupción del embarazo.
Pronóstico.- Por lo general, el LMPS es letal durante el segundo o el tercer trimestre de gestación.
Bibliografía
- Genetics home reference. Multiple pterygium syndrome.:http://ghr.nlm.nih.gov/condition/multiple-pterygium-syndrome. Nov. 2011
- Wallis CE., Shun-Shin M, and Beighton PH- (1988). Autosomal dominat antecubital pterygium: Syndrome status substantiated. Clin.Genet. 34,64-69.
- Bussiere JA. Developement abnormal d’un faisceau musculare acromio-mastoidien rudimentaire, malformation congenitale rare, observe’e a pondicherry (Indes Orientales). Annales d’hygiene et de medecine coloniales 1902;5:686-
- Frawley JM. Congenital webbing. Am J Dis Child 1925;29:799-805.
- Escobar V, Bixler D, Gleiser S, Weaver DD, Gibbs T. Multiple pterygium syndrome. Am J Dis Child 1978;132:609-61. Vogt J, Harrison BJ, Spearman H, Cossins J, Vermeer S, ten Cate LN, et al. Mutation analysis of CHRNA1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet 2008; 82(1): 222-7.
- Jones KL. Smith’s recognizable patterns of human malformation. Philadelphia: Saunders, 2006.
- Escobar V, Bixler D, Gleiser S, Weayer DD, Gibbs T. Multiple pterigium syndrome. Am J Dis Child 1978;132: 609-11.
- Cruz M, Bosh J. Atlas de malformaciones congénitas. Barcelona: Espaxs, 1998.
- Camacho Franco L, Nualart L, Capdevila LR. Síndrome de Escobar o pterigium multiple congénito. Reporte de caso. Acta Ortoped Mex 2006; 214
- Ramer JC, Ladda RL, Demuth WW. Multiple pterigium syndrome. An overview. Am J Dis Child 1988; 142 (7): 794-8.
- Hoffmann K, Muller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, et al.. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet 2006; 79(2):
- Morgan NV, Brueton LA, Cox P, Greally MT, Tolmie J, Pasha S, et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterigium syndrome. Am J Hum Genet 2006; 79: 390-5.
- Chen H, Immken L, Lachman R, Yang S, Rimoin DL, Rightmire D, et al. Syndrome of multiple pterygia, camptodactyly, facial anomalies, hypoplastic lungs and heart, cystic hygroma, and skeletal anomalies: delineation of a new entity and review of lethal forms of multiple pterygium syndrome. Am J Med Genet 1984; 17: 809-26.
- Chen H, Chang CH, Misra R, Peters H, Grijalba N, Opitzj. Multiple pterigium syndrome. Am J Med Genet 1980; 7: 91-102.
- McCall RE, Budden J. Treatment of multiple pterigium syndrome. Orthopedics 1992; 15(7): 1417-22.
- Vogt J.CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J Med Genet. 2012 Jan;49(1):21-6.
- Robison KG Sinapsis neuromotor en el síndrome de Escobar. Am J Med Genet A. 2013 Dic; 161A (12): 3042-8
- Kodaganur SG Fenotipo clínico y la ausencia de mutaciones en los genes CHRNG, CHRND y CHRNA1 en dos familias indias con síndrome de Escobar. Clin Dysmorphol. 2013 Apr; 22 (2): 54-8.