INFORMACIÓN BÁSICA.- SÍNDROME DE DELECION 22q. Microdeleción 22q11 (OMIM: 188400-192430).– La microdelecion 22q11 es una deleción genética frecuente con variabilidad fenotípica amplia. Engloba una serie de síndromes, entre los que destaca el síndrome de DiGeorge.
La mayoría de los pacientes afectados por el síndrome de deleción 22q11 no experimentan todos los problemas de salud que se asocian con él. En general los problemas que están presentes debido al síndrome de deleción 22q11 se puede controlar con tratamiento, especialmente si se detecta a tiempo.
La microdeleción 22q11 está implicada en una serie heterogénea de fenotipos, como los síndromes de DiGeorge, CATCH 22, el cardiofacial de Cayler y otros. Esta anomalía constituye la alteración cromosómica más frecuente (1:4.000 recién nacidos) en el ser humano, siendo el 93% de los casos mutaciones de novo.
Las manifestaciones clínicas más frecuentes que expresan estos síndromes destacan: malformaciones cardiacas, microcefalia y facies característica: pabellones auriculares pequeños y contorno superior recto, fisuras palpebrales antimongoloides y telecantus, disfunción velopalatina, con o sin paladar hendido, y labio leporino.
También puede asociarse a hipoparatiroidismo, inmunodeficiencia por hipoplasia tímica, retraso psicomotor y alteraciones psiquiátricas en la edad adulta.
Los signos y síntomas del síndrome de deleción 22q11.2 pueden variar de un paciente afectado a otro. Por esta razón, varios trastornos y las colecciones de los síntomas causados por este síndrome tienen otros nombres.
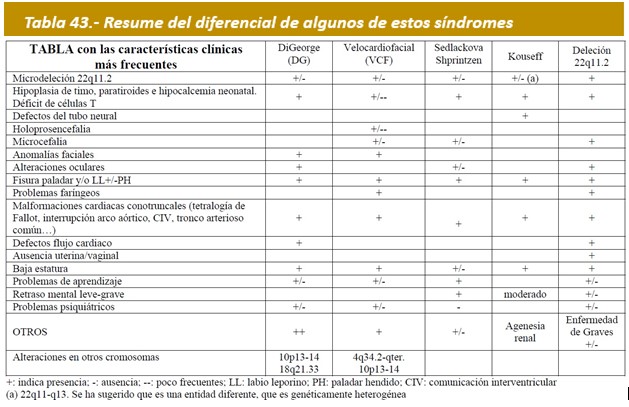
Estos nombres, algunos de los cuales todavía se utilizan en ocasiones, incluyen las siguientes entidades sindrómicas; la Tabla 43 resume las características diferencias de algunas de ellas. Cada una de ellas caracterizadas:
- – El síndrome de DiGeorge
- – Secuencia de DiGeorge
- – El síndrome velocardiofacial (VCFS)
- – El síndrome de Sedlackova -Shprintzen
- – El síndrome de anomalías conotruncales y de la cara (CTAF)
- – El síndrome cardiofacial de Cayler
- – El síndrome CATCH- 22
- – El síndrome de Takao
Además, algunos niños con el síndrome de deleción 22q11.2 han sido diagnosticados previamente con el síndrome Opitz G / BBB (ligado al cromosoma X), o síndrome cardiofacial de Cayler. Los médicos ahora entendemos que estos trastornos comparten la misma causa genética como el síndrome de deleción 22q11.2.
Prevalencia: Aproximadamente uno de cada 4.000 personas tienen el síndrome de deleción 22q11.2. Algunos expertos creen que este número debería ser mayor porque algunas personas con el defecto de cromosomas tienen síntomas menos severos que no pudieron ser diagnosticados como síndrome de deleción 22q11.2. Por lo tanto, no saben que tienen el síndrome.
Causas: A la mayoría de los pacientes con síndrome de deleción 22q11.2 les falta alrededor de 30 a 40 genes. La función exacta de muchos de estos genes sigue siendo un misterio. Pero un gen, TBX1, probablemente explica los síntomas físico más común del síndrome, incluyendo problemas del corazón y el paladar hendido.
Otro gen cercano, llamado COMT, también puede ayudar a explicar el mayor riesgo de problemas de conducta y las enfermedades mentales en las personas con el síndrome.
Alrededor del 90% de los casos del síndrome de supresión 22q11.2 se producen al azar en la fertilización o temprano en el desarrollo fetal. Así que la mayoría de pacientes afectados con este trastorno no tienen antecedentes familiares de la misma. Sin embargo, pueden pasar la enfermedad a sus hijos. El restante 10% de los casos se hereda de la madre o el padre. Cuando la condición es hereditaria, otros miembros de la familia también podrían verse afectadas.
Debido a que una persona que tiene esta supresión cromosómica tiene una probabilidad del 50% de los que pasa la supresión a un niño, generalmente se les ofrece a ambos padres la oportunidad de un análisis de sangre para buscar la eliminación.
Manifestaciones clínicas: Los signos y síntomas del síndrome de deleción 22q11.2 pueden variar ampliamente, incluso entre los miembros de la familia. Por lo menos 30 diferentes signos o síntomas han sido asociados con este trastorno, pero la mayoría de las personas afectadas sólo tienen algunos síntomas.
Los síntomas más comunes incluyen: defectos del corazón, que habitualmente están presentes desde el nacimiento, anomalías del paladar, como el paladar hendido y dificultades en el habla, infecciones del oído medio o la pérdida de audición, dificultades para la alimentación, niveles bajos de calcio en la sangre, debido a problemas con las glándulas paratiroides, que pueden desencadenar crisis de hipocalcemia, problemas del sistema inmune que puede aumentar el riesgo de infecciones, anomalías renales, insuficiencia respiratoria de causa multifactorial debido a las malformaciones de la vía aérea, aunque en la mayoría de los pacientes son leves, por lo que no suelen producir sintomatología.
Sin embargo, en algunos casos pueden condicionar clínica respiratoria grave, problemas de columna vertebral, como escoliosis y anormalidades en los huesos del cuello o superior de la espalda, dificultades de aprendizaje, especialmente con material no verbal, y retrasos en el desarrollo y comunicación, así como problemas de interacción social, incluyendo el autismo, un mayor riesgo para las enfermedades mentales, como ansiedad, depresión o esquizofrenia en la edad adulta.
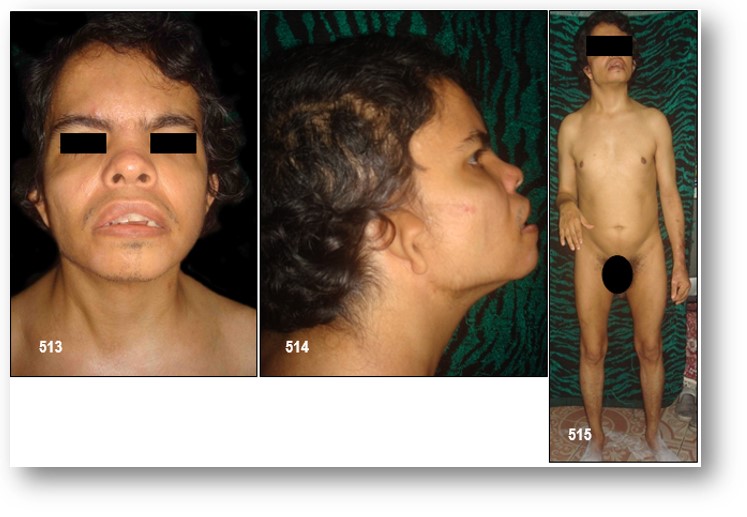
Las características faciales de los niños con síndrome de deleción 22q11.2 pueden incluir los siguientes: orejas pequeñas con forma cuadrada en la parte superior, párpados encapuchados, labio y/o paladar leporino, imagen facial asimétrica al llorar, pequeña boca y entre abierta, mentón y los lados de la punta de la nariz.
Los síntomas del síndrome de deleción 22q11.2 pueden parecerse a otras condiciones o problemas médicos.
Observaciones recientes indican que el tamaño de la deleción y la modificación de lugares genéticos no son responsables de todas las diferencias fenotípicas observadas hasta ahora (CATCH22). Además no ha sido encontrada la deleción 22q11 en un defecto cardiaco aislado. Existe el reporte de un caso clínico en donde el niño afectado contaba con la citogenética y fenotipo normal de DiGeorge, y el padre portador era asintomático con un cariotipo mosaico con una supresión 22q11 en tres diferentes líneas celulares.
Diagnóstico: El diagnóstico se sospecha tras un examen clínico-semiológico y la detección de anomalías (defectos cardíacos por ecocardiografía, anomalías vertebrales por rayos X de la columna cervical). Se confirma por la detección de la deleción 22q11.2, usando FISH, MLPA, aCGH o microarrays SNP de genoma completo.
El diagnóstico prenatal es posible en casos familiares por amniocentesis o biopsia corial, y en embarazadas donde se hayan observado anomalías asociadas por ecocardiografía fetal. Es posible el diagnóstico genético preimplantacional.
La deleción surge de novo en el ~90% de los casos. Hay un riesgo de recurrencia del 50% en los individuos afectados.
Diagnóstico diferencial.- El diagnóstico diferencial incluye los síndromes de Smith-Lemli-Opitz, CHARGE, de Alagille, VATER, de Goldenhar y la embriopatía por isotretinoina.
Tratamiento: No hay cura para el síndrome de deleción 22q11.2, pero muchos de sus problemas de salud relacionados pueden ser tratados. El tratamiento específico para el síndrome de deleción 22q11.2 será determinado por el médico de su hijo basándose en lo siguiente: la edad de su hijo, estado general de salud y sus antecedentes médicos la extensión de la enfermedad el tipo de enfermedad la tolerancia de su hijo a determinados medicamentos, procedimientos o terapias las expectativas para la trayectoria de la enfermedad su opinión o preferencia.
El tratamiento también dependerá de las características particulares de cada niño, que pueden incluir los siguientes:
Los defectos cardíacos serán evaluados por un cardiólogo y se puede corregir con cirugía o procedimiento intervencionista.
Un cirujano plástico y un fonoaudiólogo evaluarán el labio leporino y/o paladar defectos.
Especialistas del habla y el sistema gastrointestinal evaluarán las dificultades de alimentación. Algunos niños con este problema tienen graves dificultades de alimentación y la necesidad de alimentación por sonda a fin de obtener una nutrición adecuada. Las dificultades para alimentarse, no suelen estar relacionados con problemas cardíacos o paladar, sino con los problemas intestinales causados por el síndrome.
Exámenes inmunológicos se debe realizar en todos los niños con esta supresión. Para controlar el trastorno de células T y las infecciones recurrentes, las vacunas de virus vivos deben evitarse y todos los productos de sangre para transfusiones (si es necesario) deben ser irradiados a menos que sean analizados por un médico inmunólogo.
Otros problemas comunes que pueden requerir tratamiento incluyen: calcio bajo. Esto es común en los niños con el síndrome, en especial inmediatamente después del nacimiento. Pero también se puede recurrir en los períodos de estrés, como durante la pubertad o después de la cirugía. Un niño puede necesitar tomar suplementos de calcio y vitamina D para ayudar a absorber el calcio. Una referencia a un endocrinólogo (médico que se especializa en el tratamiento de condiciones que afectan al sistema endocrino puede ser recomendada. dificultades en el desarrollo.
Los niños pequeños con síndrome de deleción 22q11.2 pueden ser lentos para alcanzar los hitos del desarrollo como sentarse, caminar y hablar. El Organismo Internacional de la Fundación Síndrome de Supresión 22q11.2 recomienda que los padres consideran la terapia física (PT), terapia ocupacional (TO), y terapia del habla para sus hijos afectados. PT fortalece los músculos grandes y ayuda a los niños superarán las etapas de desarrollo. OT se centra en los músculos pequeños utilizados para las habilidades tales como atarse los zapatos y la ropa abotonar. También puede ayudar con problemas de alimentación. La terapia del habla puede ayudar a resolver retrasos en el lenguaje que pueden aparecer tras el niño cumple 1 año de edad.
Perspectivas a largo plazo para los niños con síndrome de deleción 22q11.2: Un pequeño porcentaje de niños con defectos cardíacos y problemas graves del sistema inmunológico causado por el síndrome de deleción 22q11.2 no sobrevivirán el primer año de vida. Sin embargo, con el tratamiento adecuado para los defectos cardíacos, los trastornos del sistema inmunológico y otros problemas de salud, la gran mayoría de niños con deleción 22q11.2 sobrevivirán y podrán desarrollarse en la edad adulta. Generalmente, estos niños necesitarán ayuda extra en la escuela y un cuidado a largo plazo para sus necesidades de salud individuales.
Aunque no es común, algunos niños con esta enfermedad pueden tener condiciones de comportamiento como el autismo, el trastorno por déficit de atención (ADD/ADHD), trastorno obsesivo compulsivo (TOC), o la ansiedad. De acuerdo con el Velo-cardio-facial, la mayoría de las personas con este problema tienen algunos problemas de conducta. La aparición de los trastornos psiquiátricos es más alta para los adultos con síndrome de deleción 22q11.2, pero los investigadores médicos están aún investigando la naturaleza exacta y el riesgo de estos trastornos. A continuación describe en detalles cada unos de los síndromes relacionados con la deleción del cromosoma 22q11.2.
Pronóstico.- El pronóstico es variable y depende de la gravedad de la enfermedad. La tasa de mortalidad infantil es relativamente baja (~4%); en adultos, la mortalidad es mayor que en el resto de la población adulta.
Nos referiremos exclusivamente al Síndrome de DiGeorge.
SÍNDROME DE DIGEORGE (APLASIA O HIPOPLASIA TÍMICA). (OIM 182400; 192430).- El síndrome de DiGeorge es una hipoplasia o aplasia tímica y paratiroidea que da lugar a una inmunodeficiencia del linfocito T y un hipoparatiroidismo.
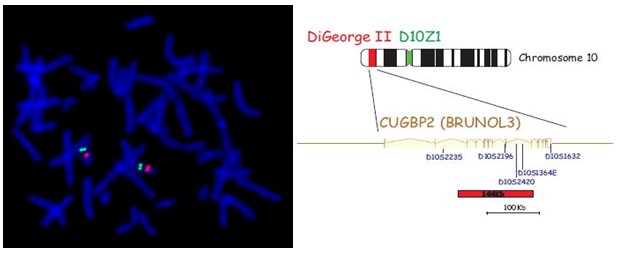
El síndrome de DiGeorge es un trastorno de inmunodeficiencia primaria con defectos de células T. Se debe a deleciones o supresiones génicas en la región cromosómica de DiGeorge en 22q11, mutaciones en otros genes del cromosoma 10p13 y mutaciones en otros genes desconocidos, lo que causa una disembriogenia de estructuras que se desarrollan a partir de las bolsas faríngeas durante la 8ª semana de gestación. La mayoría de los casos son esporádicos; se ven igualmente afectados niños y niñas. La herencia es autosómica dominante.
El síndrome de DiGeorge puede ser:
- Parcial: las células T funcionan en cierto grado
- Completa: la función de las células T está ausente.
Los lactantes con síndrome de DiGeorge tienen pabellones auriculares de implantación baja, hendiduras faciales en la línea media, una mandíbula pequeña y retraída, hipertelorismo, surco nasolabial acortado, retraso del desarrollo y cardiopatías congénitas. El diagnóstico se basa en los hallazgos clínicos e incluye una evaluación de la función inmunitaria y paratiroidea, así como análisis cromosómico. Tratamiento: incluye medidas sintomáticas y, si es grave, trasplante de células del timo de células madre.
Sinónimos: Tercera y Cuarta Bolsa Faríngea, Síndrome de la Agenesia Tímica Aplasia Tímica, Tipo Di GeorgeHarrington, Síndrome de Hipoplasia Tímica, Tipo Di George Bolsa Faríngea, Síndrome de la Aplasia Congénita del Timo.
Acrónimo: CATCH 22 (Cardiac defects, Abnormal facial features, Thymic hypoplasia, Cleft palate and Hypocalcemia).
Se ha observado suficiente heterogeneidad en la expresión fenotípica de pacientes con la deleción 22q11.2.
Parece no existir un claro acuerdo en su nomenclatura, ya que hay múltiples denominaciones. Más bien se asume que son diferentes expresiones de la misma alteración.
Definición: El Síndrome de DiGeorge se considera una anomalía localizada de expresión múltiple, que puede presentarse aislada o formando parte de cuadros polimalformativos sindrómicos.
El síndrome de DiGeorge es una enfermedad rara del desarrollo, caracterizada por tetania (espasmos dolorosos o de torsión, incontrolables que atenazan e impiden el movimiento normal), enfermedad cardiaca al nacimiento, cara peculiar, mayor frecuencia de infecciones y ausencia o escaso desarrollo del timo y de las glándulas paratiroides; lo que da lugar a inmunodeficiencia de células T e hipocalcemia.
Estos defectos ocurren en la tercera y la cuarta bolsa faríngea. Es frecuente la afección de otras estructuras que se desarrollan en el mismo período; y se producen anomalías en los grandes vasos, dextroposición del arco aórtico, cardiopatías congénitas como son los defectos de los tabiques auricular y ventricular, atresia esofágica, úvula bífida, filtrum ancho, hipertelorismo, canto externo del ojo más bajo que el canto interno, hipoplasia mandibular y orejas de implantación baja y a menudo hendidas.
La alteración del timo genera un déficit de la inmunidad celular, estando intacta la inmunidad humoral. Otros aspectos aparte de los defectos en el tracto eferente del corazón, existen anomalías craneofaciales (síndrome velocardiofacial).
Según el grado de aplasia tímica, el síndrome puede ser parcial o completo. En el grado parcial los niños tienen problemas de infecciones de repetición, aunque no se afecte el desarrollo. En el grado completo los pacientes tienen una gran susceptibilidad a infecciones por microorganismos oportunistas, como los hongos, los virus y Pneumocystis jiroveci.
El síndrome de DiGeorge no es propiamente un síndrome sino una anomalía localizada de expresión múltiple, que puede presentarse aislada o formando parte de cuadros polimalformativos sindrómicos o no (síndrome es el patrón de múltiples anomalías que afectan a múltiples áreas del desarrollo y que tienen etiopatogenias, causas y mecanismos de producción de enfermedad, relacionadas).
Desde el punto de vista genético y sindromático; es consecuencia de una mutación en el gen TBX1 por una microdeleción submicroscópica intersticial en 22q11.21- 11.23 cromosómica a nivel del cromosoma 22; es el síndrome de microdeleción más común.
El síndrome de DiGeorge se produce por igual en ambos sexos; se estima una prevalencia de alrededor de 1 por cada 3,000 a 4,000 nacimientos vivos aproximadamente; se transmite entre familiares de manera autosómica dominante en 8 a 25% de los casos (alteraciones genéticas tipo mosaicismo); los padres pueden estar completamente asintomáticos. Esta trasmisión familiar es bastante rara o de una mutación tipo de novo» que es mucho más común.
Los pacientes portadores de esta microdeleción nacen normalmente con anomalías conotroncales e inmunodeficiencia leve a moderada. Todavía no se entiende con certeza el porqué de la enorme heterogenicidad fenotípica de este síndrome. La microdeleción 22q11 es la causa de muchos síndromes clínicos, entre ellos el síndrome Velocardiofacial y el síndrome de DiGeorge.
Historia: La historia del síndrome de Di George incluye los siguientes descubrimientos: A mediados de la década del 60, el médico endocrinólogo Angelo Di George detectó que un grupo particular de características clínicas con frecuencia se presentaban juntas. Estas características incluían: Hipoparatiroidismo, trastorno que produce hipocalcemia. Timo hipoplásico o ausencia de timo, trastorno que produce problemas en el sistema inmunológico. Defectos cardíacos conotroncales (es decir, tetralogía de Fallot, cayado aórtico interrumpido, comunicación interventricular, anillos vasculares). Labio leporino y/o paladar hendido. En la década del 70, el Dr. Robert Shprintzen, patólogo del habla, describió a un grupo de pacientes con características clínicas similares, incluyendo labio leporino y/o paladar hendido, defectos cardíacos conotroncales, ausencia de timo o timo hipoplásico e hipocalcemia (en algunos casos). El Dr. Shprintzen llamó a este grupo de características síndrome velocardiofacial, aunque también se conoció como síndrome de Shprintzen. En la década del 80, se desarrolló una tecnología capaz de identificar un defecto cromosómico subyacente en estos síndromes. Se determinó que más del 90% de todos los pacientes que presentaban las características de los síndromes de Di George y Shprintzen (síndrome velocardiofacial) tenían una deleción cromosómica en la región 22q11.
En otras palabras, se trataba del mismo síndrome que, al ser descrito por diversos investigadores de diferentes especialidades, había recibido varios nombres. En la actualidad, muchos médicos e investigadores utilizan el término síndrome de deleción 22q11.2 porque el mismo describe el problema cromosómico subyacente o bien el término síndrome velocardiofacial (velo-cardio-facial syndrome, VCFS) porque describe los principales sistemas del cuerpo. A efectos del contenido de esta publicación, lo denominaremos síndrome de DiGeorge.
Etiopatogenia: El 90% de los pacientes con las características de este síndrome carecen de una pequeña parte del cromosoma 22 en la región q11. Esta región abarca cerca de 30 genes individuales y es responsable de defectos del desarrollo en estructuras específicas de todo el cuerpo.
No se sabe por qué esta región del cromosoma 22 es más propensa a ser eliminada, pero se trata de uno de los defectos cromosómicos más frecuentes en los recién nacidos. Se estima que la deleción 22q11 se produce en uno de cada 3000 a 4000 recién nacidos vivos. La mayoría de los casos de deleción 22q11 son episodios nuevos o esporádicos (aparecen por casualidad). Sin embargo, en un 10% de las familias, la deleción es hereditaria y afecta o podría afectar a otros miembros de la familia que corren el riesgo de transmitir esta enfermedad a sus hijos. Éste es un gen autosómico dominante, por lo que cualquier persona que sufra esta deleción tiene un 50% de probabilidades de transmitirla a alguno de sus hijos. Por esta razón, siempre que se diagnostica una deleción, se ofrece a ambos padres la oportunidad de realizarse un análisis de sangre para buscar y detectar dicha deleción.
Aproximadamente un 10% de las personas que tienen las características del síndrome velocardiofacial (VCFS) no sufren una deleción en la región q11 del cromosoma 22. Se han asociado otros defectos cromosómicos a estas características, como la diabetes gestacional, el síndrome de alcoholismo fetal y la exposición prenatal al Accutane® (un medicamento para el acné quístico).
La alteración genética responsable se ha hallado en la deleción de la región 22q11.2, por lo que actualmente se engloba dentro del grupo de trastornos del síndrome de deleción 22q11. Principalmente se describen malformaciones cardíacas, inmunodeficiencia, hipocalcemia, malformaciones en paladar con insuficiencia velofaríngea, dificultad para el aprendizaje y retraso del desarrollo. Otras asociaciones menos frecuentes son las malformaciones genitourinarias, enfermedad psiquiátrica y alteraciones osteoarticulares.
Manifestaciones clínicas: Dentro de las características clínicas más comunes del síndrome de DiGeorge, se pueden incluir: Un 69 por ciento de niños con anormalidades en el paladar (como labio leporino y/o paladar hendido). Un 30% con dificultades de alimentación. Un 80% con defectos cardiacos conotroncales.
Los defectos cardíacos que se asocian más comúnmente a este síndrome son las anomalías del arco aórtico (arco aórtico a la derecha, doble, o interrupción del arco aórtico tipo B), comunicación interventricular mal alineada, atresia o estenosis pulmonar, tetralogía de Fallot y tronco arterioso.
La tetralogía de Fallot es la presentación más común entre las cardiopatías congénitas cianóticas, con una incidencia de 0.5:1,000. Un 40% con pérdida de la audición o exámenes de oído anormales. Un 30% con anomalías genitourinarias (agenesia renal unilateral, o malformación renal, hidronefrosis, quistes renales; hipospadias, criptorquidia, hernias umbilicales e inguinales; ageneseia uterina). Un 60% con hipocalcemia. Un 40% con retardo mental (generalmente de mínimo a leve). Cocientes de inteligencia (IQ) que generalmente se encuentran en un rango de 70 a 90. Un 90% con dificultad en el aprendizaje. Un 70% con disfunciones inmunológicas graves (el sistema inmunológico no funciona correctamente debido a linfocitos T anormales, provocando frecuentes infecciones). Anomalías oculares: opacidad corneal, microtalfmia, coloboma del iris. Detención del crecimiento que sin embargo a la edad adulta refleja la media estatura de padres y hermanos; sobrepeso sobre todo a la edad adolescente. La Tabla 39 resume lo descrito.
Entre otras manifestaciones se incluyen: deficiencias hormonales (hormona del crecimiento), infecciones recurrentes. Bajo peso al nacer, dedos largos y delgados (a veces manos grandes), e incluso características mucho más raras.
Entre las manifestaciones odontológicas se citan: Retardo en la formación y erupción de la dentición permanente, cares dentales debido a anomalías del esmalte y al carácter quebradizo de los dientes; maloclusión.
Las características faciales pueden incluir: orejas pequeñas y cuadradas en la parte superior, párpados caídos, labio leporino y/o paladar hendido, facies de llanto asimétricas, boca, mentón y zonas laterales de la punta de nariz de tamaño reducido, pómulos prominentes. Los síntomas del síndrome de DiGeorge pueden parecerse a los de otros trastornos o problemas médicos.
Parece ser que el fenotipo depende de diferentes variables basadas en los cambios en diferentes locus.

Diagnostico: Los pacientes con síndrome de DiGeorge suelen diagnosticarse en edad pediátrica por cardiopatía congénita, hipocalcemia sintomática, infecciones recurrentes o no habituales, y/o fenotipo característico.
Hasta aquí se puede deducir que nos hallamos dentro del 40% de los pacientes sin alteración manifestó del calcio. Las alteraciones neurológicas aparecen en un 8%, y se asume que la hipocalcemia es el agente causal en el 68% de los pacientes con crisis epilépticas.
El estándar de oro para diagnosticar las alteraciones a nivel cromosómico es la hibridación de la fluorescencia in situ (HFIS); sin embargo, la clínica es de gran utilidad en estos casos, ya que estos pacientes presentan rasgos característicos de la enfermedad.
Al cuadro clínico del síndrome de DiGeorge lo podemos clasificar en signos y síntomas mayores y menores, dependiendo de la frecuencia con que se presentan:
Mayores:
- – Anomalías cardiacas 75%.
- – Tetralogía de Fallot 49-83%.
- – Arco aórtico interrumpido 17-22%.
- – Defecto del septum ventricular 14-15%.
- – Tronco arterioso 13-14%.
- – Cuenta baja de linfocitos T 75-80%.
- – Hipocalcemia 17-60%.
- – Alteraciones en el paladar 69-100%.
- – Problemas de lenguaje 79-84%.
Menores:
- – Alteraciones oftalmológicas 7-70%.
- – Alteraciones renales 36-37%.
- – Alteraciones en el comportamiento 9-50%.
Criterios Diagnóstico para Síndrome DiGeorge
Diagnóstico Definitivo
Paciente hombre o mujer con recuento reducido de células T CD3+ (menos de 500/mm3) y dos de las 3 características siguientes:
- – Defecto cardíaco conotruncal (tronco arterioso, tetralogía de Fallot, arco aórtico interrumpido, o subclavia derecha aberrante).
- – Hipocalcemia de más de 3 semanas de duración que requiere tratamiento.
- – Deleción del cromosoma 22q11.2
Diagnóstico Probable
Paciente hombre o mujer con recuento reducido de células T CD3+ (menos de 1500/mm3) y la característica siguiente:
– Deleción del cromosoma 22q11.2
Diagnóstico Posible
Paciente hombre o mujer con recuento reducido de células T CD3+ (menos de 1500/mm3) y al menos uno de los siguientes:
- – Defecto cardíaco
- – Hipocalcemia de más de 3 semanas de duración que requiere tratamiento
- – Fascie dismorfica o anormalidad palatina
Los pacientes con diagnóstico definitivo o probable son asumidos que tiene más del 98 y 85 por ciento de probabilidad, respectivamente, que en 20 años tendrán el mismo diagnóstico. Los pacientes con un diagnóstico posible son aquellos que tendrán algunas pero no todas las características clínicas o hallazgos de laboratorio de una enfermedad en particular.
Además de la historia prenatal, realizar los trámites de historial médico y familiar, y un examen físico, diagnosticar el síndrome de deleción 22q11.2 pueden incluir: análisis de sangre y estudios para determinar problemas del sistema inmunológico rayos X – examen de diagnóstico que utiliza energía electromagnética invisible para obtener imágenes de tejidos internos, huesos y órganos en una placa. Ecocardiografía. La hibridación fluorescente in situ (FISH) estudios – cuando las características de los defectos cardíacos conotruncales, hendiduras, otras características faciales, hipocalcemia y ausencia del timo son identificados, un análisis de sangre generalmente se indica a buscar una deleción en el cromosoma región 22q11.2.
FISH está diseñado específicamente para buscar pequeños grupos de genes que se eliminan. Si el FISH no detecta ninguna supresión en la región 22q11.2 y las características del síndrome velocardiofacial son muy evidentes, a continuación, un estudio completo de los cromosomas se realiza generalmente en busca de otros defectos cromosómicos que se han asociado con este síndrome.
Si una deleción 22q11.2 se detecta en un niño, ambos padres se les ofrece la prueba FISH para ver si ésta es hereditaria. En aproximadamente el 10 por ciento de las familias, la supresión ha sido heredada de uno de los padres. Cualquier persona que presente esta supresión 22q11.2 tiene una probabilidad del 50 por ciento, en cada embarazo, de transmitirla a un hijo.
FISH: La aplicación de la técnica de FISH para identificar la deleción 22q11.2 en pacientes con fenotipo de SD22q11, ha permitido concluir que los fenotipos reportados como: VCFS, secuencia de Di George, síndrome de Sedlačková, síndrome de anomalías conotruncales y de la cara, síndrome de Cayler y CATCH 22, son descripciones de la misma patología en una entidad clínica que expresa un fenotipo variable.
El marcador molecular de este síndrome es la microdeleción hemizigota del segmento q11.2 en el brazo largo del cromosoma 22.
Esta deleción es una de las anomalías cromosómicas más comunes, con una frecuencia de 1:2000 a 1:4000 en la población general. Un 85-90% de los casos con SD22q11.2 tiene un deleción típica de ~3 millones de pares de bases, que incluyen ~40 genes, 10-12% tienen una deleción de ~1,5 millones de pares de bases y unos pocos muestran pequeñas deleciones. La mayoría de los casos son deleciones de novo, sin embargo, un 5-15% de los pacientes muestran una enfermedad autosómica dominante.
La presencia de dismorfismo o cardiopatía congénita cianógena con una anomalía conotruncal en los tres casos reportados, indujo a efectuar el estudio citogenético que permitió demostrar la deleción diagnóstica en la banda q11.2 del cromosoma 22.
SD22q11 (VCFS) tiene un amplio espectro fenotípico, con hallazgos en el examen físico y manifestaciones cardiológicas, metabólicas, endocrinas y del comportamiento. Se han reportado más de 180 características, las cuales se enumeran en la publicación de la “VCFS Educational Foundation”. Con frecuencia se asocian con el síndrome: paladar hendido (>8%) y anomalías congénitas cardiacas conotruncales (15% de las Tetralogías de Fallot y 50% de las interrupciones del arco aórtico tipo B y tronco arterial común). Se reporta una frecuencia de cardiopatía congénita del 71% y de alteraciones del paladar del 75%. Sin embargo, las características más frecuentes son las alteraciones del comportamiento y cognitivas. Las anomalías estructurales más frecuentes son las vasculares, como localización aberrante y la ausencia o presencia de pequeños vasos sanguíneos. Se ha definido además un dismorfismo facial característico .
Los pacientes que tienen la secuencia secundaria de Di George (SDG) presentan también una facies característica, tetania por hipocalcemia, inmunodeficiencia congénita de la célula T e hipoplasia de timo. Además, la anatomía patológica muestra defectos en las glándulas paratiroides, en el arco aórtico y cardiaco.
El espectro de la inmunodeficiencia varía entre una inmunodeficiencia severa y un sistema inmune normal. En dos de los casos reportados no se realizó un estudio inmunológico; en el otro se demostró una inversión de la tasa CD4/CD8 asociada a infección recurrente de vías respiratorias, pero no se encontró hipocalcemia, por lo que se debe concluir que entre los casos reportados solo se puede sospechar un caso de SD22q11 con SDG. El manejo de los casos se basa en el tratamiento médico quirúrgico de la cardiopatía y del paladar hendido, así como en la atención de los problemas de comportamiento, cognitivos y de aprendizaje.
La disponibilidad de la técnica de FISH para identificar la microdeleción 22q11.2, permite un diagnóstico citogenético molecular de los SD22q11, y la posibilidad de que reciban un manejo sindrómico más allá del tratamiento de la cardiopatía y el paladar hendido, lo cual posibilitará que estos niños disfruten de una mejor calidad de vida al recibir un abordaje interdisciplinario que incluya las áreas escolar y del comportamiento.
Aproximadamente el 90% de los pacientes con síndrome de DiGeorge o VCFS tendrá una prueba de FISH positivo. Para aquellos con una prueba normal, un porcentaje pequeño puede tener una deleción que afecta al brazo corto del cromosoma 10 (que puede ser probado con una prueba de FISH diferente), pero la mayoría no tienen anomalía cromosómica que se puede encontrar actualmente. Si los médicos que atienden a su hijo a hacer el diagnóstico de síndrome de DiGeorge o VCFS sobre la base de ciertas características típicas (apariencia facial, enfermedades del corazón, etc.), luego de que sigue siendo cierto incluso si las pruebas FISH son normales.
Genética Molecular: Genes en la deleción: Se han identificado numerosos genes dentro de la región deletada de manera más común en 22q11.2.
En su búsqueda por genes, los investigadores también han buscado genes que pudieran tener un papel en el desarrollo de los arcos branquiales o la cresta neural. Varios genes candidatos han recibido atención particular (IDD/SEZI/LAN, GSCL, HIRA, UFD1L), pero todos resultaron negativos para mutaciones en pacientes con VCFS sin una microdeleción 22q11. COMT, el gen que codifica para la catecol-O-metil transferasa, tiene un papel crucial en el metabolismo del neurotransmisor dopamina. Se cree que la función anormal de las vías dopaminérgicas juega un papel importante en la esquizofrenia. Dado que el gen que codifica para la COMT se localiza en 22q11, se le considera un candidato primario para la etiología de la esquizofrenia en el VCFS. Se ha sugerido que el polimorfismo genético funcional común del gen COMT, que da como resultado diferencias de actividad de entre 3 a 4 veces podría contribuir a la etiología de desórdenes psiquiátricos. Dos estudios reportaron que en una población de pacientes con VCFS, hay una asociación aparente entre el alelo de baja actividad, COMT158met en el cromosoma no deletado y el desarrollo de un desorden de espectro bipolar y, en particular, una forma cíclica rápida.
Tratamiento: El tratamiento específico del síndrome de Di George será determinado por el médico de su hijo basándose en lo siguiente: la edad de su hijo, su estado general de salud y su historia médica; qué tan avanzada está la enfermedad; el tipo de enfermedad; la tolerancia de su hijo a determinados medicamentos y procedimientos o a terapias específicas; sus expectativas para la evolución de la enfermedad. El tratamiento también dependerá de las características específicas de cada niño en particular y puede incluir lo siguiente:
Los defectos cardiacos serán evaluados por un cardiólogo. Un cirujano plástico y un patólogo del habla evaluarán el labio leporino y el paladar hendido. Los especialistas gastrointestinales y del habla evaluarán las dificultades de alimentación. Se realizarán evaluaciones inmunológicas en todos los niños que padezcan esta deleción.
Para controlar el trastorno de los linfocitosT y las infecciones recurrentes, se debe evitar la aplicación de vacunas con virus vivos y todos los productos derivados de la sangre, para la realización de transfusiones (si fueran necesarias) deberán irradiarse, a menos que los mismos sean aprobados por un médico especialista en inmunología. El síndrome de Di George es un trastorno genético con cuadros variables presentes en cada persona que lo padece. Sin embargo, las condiciones que son comunes del síndrome incluyen determinados defectos cardiacos, efectos en el aspecto del rostro y ausencia o subdesarrollo del timo y las glándulas paratiroides.
Un porcentaje de niños con defectos cardíacos y problemas graves del sistema inmunológico no sobreviran el primer año de vida. Sin embargo, aplicando el tratamiento adecuado para los defectos cardíacos, los trastornos del sistema inmunológico y otros problemas de salud, la gran mayoría de niños con deleción 22q11.2 sobrevivirán y podrán desarrollarse. Generalmente, estos niños necesitarán ayuda extra en el período escolar y un cuidado a largo plazo para sus necesidades individuales. En todo caso, la conducta de intervención se basaría en:
- Síndrome parcial: suplementación con calcio y vitamina D
- Síndrome completo: El trasplante de células madre hematopoyéticas o de tejido tímico cultivado
En el síndrome de DiGeorge parcial, el hipoparatiroidismo se trata con calcio y vitamina D; no influye en la supervivencia a largo plazo.
El síndrome de DiGeorge completo es letal sin tratamiento, que es el trasplante de tejido tímico cultivado o de células madre hematopoyéticas. Una revisión reciente del trasplante de timo ha mostrado resultados relativamente buenos con la reconstitución de células T a los 5 a 6 meses.
Respecto al manejo quirúrgico de las anomalías cardiacas en pacientes con microdeleción 22q11, durante los últimos años se ha desatado una controversia que debate cuatro puntos del manejo:
- 1) edad ideal para la realización de la reparación total de la cardiopatía,
- 2) manejo quirúrgico en una o varias etapas,
- 3) técnica quirúrgica de acuerdo al caso y
- 4) pronóstico y supervivencia después de la reparación completa de la cardiopatía.
La literatura en general concuerda en que la reparación total de la tetralogía de Fallot es más recomendable durante el primer año de vida, si la anatomía de la cardiopatía lo permite. Es importante recalcar que la reparación temprana minimiza los efectos adversos de la hipoxia tisular y disminuye la evolución de la fibrosis e hipertrofia ventricular derecha, al igual que optimiza la función cardiaca a largo plazo, pero por otra parte aumenta la estancia postoperatoria en la Unidad de Cuidados Intensivos Neonatales o pediátricos.
Actualmente es claro que la cirugía de reparación completa en una etapa de tetralogía de Fallot es preferible en pacientes que no requieren cirugía paliativa dentro de los primeros meses de vida. La cirugía en etapas, aunque más antigua sigue siendo la recomendación en casos donde la anatomía de la cardiopatía no favorece la intervención en una sola etapa. El enfoque terapéutico que se le debe ofrecer al paciente debe ir encaminado a disminuir el riesgo acumulado que conlleva varias etapas quirúrgicas.
La técnica quirúrgica se adapta a la anatomía y condición clínica de cada paciente. Independientemente de la técnica de abordaje quirúrgico, el camino a seguir es hacia la obtención de una óptima función ventricular derecha y ampliación de su tracto de salida.
Bibliografía
- https://www.aeped.es/sites/default/files/anales/45-4-1.pdf
- DiGeorge AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. White Plains, NY: March of Dimes-Birth Defects Foundation 1968:116-21
- Di George AM. Discussions on a new concept of the cellular basis of immunology. J Pediatr. 1965;67:907-908.
- Cayler GG. Síndrome cardiopulmonar. Arch Dis Child 1969; 44 : 69 – 75.
- The Velo-Cardio-Facial Syndrome Educational Foundation. Velocardio-facial syndrome: specialist fact sheet. En: http://www.vcfsef. org/pdf/VCFS_Factsheet_07.pdf. Consultado el 28 de octubre de 2010
- International 22q11.2 deletion syndrome foundation: www 22q.org
- http://www.vcfstexas.com/index.php/support-groups/national-groups/
- Botto, L. D.; May, K.; Fernhoff, P. M.; et al.:A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 112: 101-107, 2003.
- Carelle-Calmels, N.; Saugier-Veber, P.; Girard-Lemaire, F.et al. Genetic compensation in a human genomic disorder. New Eng. J. Med. 360: 1211-1216, 2009.
- Kinouchi A, Mori K, Ando M, Takao A. Facial appearance of patients with conotruncal anomalies. Pediatr Jpn 1976; 17:84.
- Shprintzen J, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argomoso RV, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies and learning disabilities: velo-cardio-facial syndrome. Cleft Palate 1978;15:56-62.
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clínicasl features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 1997;34:798-804.
- Fagman H, Liao J, Westerlund J, Andersson L, Morrow BE, Nilsson M. The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Hum Mol Genet 2007;163:276-85. Epub 2006 Dec 12
- del Campo Casanelles, J. Pérez Rodríguez, L. García Guereta, A. Delicado, J. Quero Jiménez. CATCH-22: Implicaciones actuales de la microdeleción en 22q11. An Esp Pediatr 1996;45:341-345.
- Frutos C. Estudio de la microdeleción cromosómica en 22q11 en neonatos con cardiopatías conotruncales y del arco aórtico. RELAN 1998;1:69-73
- Driscoll DA. Prenatal diagnosis of the 22q11.2 deletion syndrome. Genet Med 2001;3:14-8.
- Davies EG, Cheung M, Gilmour K, et al: Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol140: 1660–1670.e16, 2017.