INFORMACIÓN BÁSICA.- Síndrome de Cohen (OMIM 216550).- El síndrome de Cohen (SC) es un trastorno genético del desarrollo infrecuente caracterizado por microcefalia, rasgos faciales característicos, hipotonía, déficit intelectual no progresivo, miopía y distrofia de la retina, neutropenia y obesidad truncal.
Epidemiología. – La prevalencia es desconocida. Hasta la fecha se han descrito unos 200 casos. El síndrome es más frecuente en la población finlandesa y en ciertas familias Amish, griegas/mediterráneas e irlandesas.
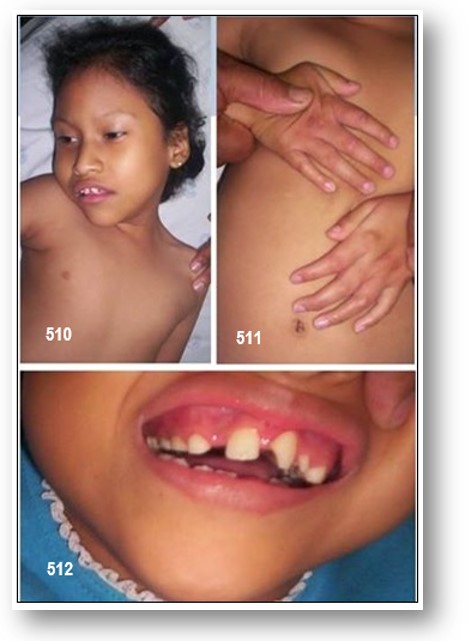
Descripción clínica.– Las manifestaciones clínicas entre familias varían. Los recién nacidos parecen normales ya que los rasgos faciales característicos todavía no están presentes, aunque la neutropenia sí puede estarlo. Las primeras manifestaciones incluyen dificultades de alimentación, hipotonía, microcefalia, retraso del desarrollo psicomotor precoz (volteo, sedestación independiente) e hipermovilidad articular. La mayoría de pacientes presentan estatura baja con manos y pies pequeños o estrechos y obesidad truncal durante la adolescencia. Los rasgos faciales (párpados ondulados o arqueados, pelo grueso, línea de implantación del cabello baja, philtrum corto, pestañas largas y gruesas, raíz nasal e incisivos centrales superiores prominentes) empiezan a aparecer alrededor de los 5 años de edad y se hacen más evidentes entre los 7-14 años y en la edad adulta.
El desarrollo del habla está retrasado. Pueden aparecer aftas. En algunos pacientes se observa infecciones respiratorias altas recurrentes (sin riesgo vital) debidas probablemente a la neutropenia. Se observa discapacidad intelectual no progresiva. Los pacientes suelen ser sociables con buen humor. En la adolescencia, se dan estrabismo y miopía junto con signos de distrofia retinocoroidea en la mayoría de casos. Con el tiempo se desarrollan nictalopía y un campo visual reducido, y la visión se deteriora progresivamente a partir de los 30 años. Los pacientes por encima de los 45 sufren de una atrofia retinocoroidea grave y cataratas subcapsulares posteriores. Aunque las anomalías visuales son graves no conllevan ceguera.
Etiología. – El SC está causado por una mutación en el gen de la vacuolar protein sorting 13B (VPS13B) en 8q22-8q23 que se piensa tiene un rol en la separación mediada por vesículas y en el tráfico intracelular de proteínas. Se han identificado más de 100 mutaciones nulas diferentes en el gen.
Métodos diagnósticos. – Para un diagnóstico definitivo del SC son necesarios los hallazgos clínicos característicos junto con las pruebas moleculares para la presencia de mutaciones en el gen VPS13B.
Diagnóstico diferencial. – Con los síndromes de Bardet-Biedl, Prader-Willi, Cri-du-chat, Alström, Angelman, Williams y MORM, y la monosomía 1p36. El síndrome de Mirhosseini-Holmes-Walton es considerado alélico del SC y es clínicamente indistinguible.
Defecto molecular.- El SC es una enfermedad genética rara; no existen estudios poblacionales de prevalencia; en la literatura solo hay alrededor de 150 casos informados en una amplia variedad de grupos étnicos y se propone que es un síndrome subdiagnosticado; la mayoría de estos pacientes son de origen finlandés y muestran un fenotipo homogéneo. Se han publicado series de casos en Bélgica, Líbano, Dinamarca e Inglaterra; en grupos humanos aislados como los Amish hay sobrerrepresentación de la enfermedad con un fenotipo muy similar al informado inicialmente en Finlandia, lo que muestra el carácter recesivo de la enfermedad. Sin embargo, el fenotipo, especialmente los rasgos faciales, varía en otros grupos étnicos, lo que dificulta el diagnóstico basado únicamente en los criterios clínicos. En Cuba y Brasil se han informado casos de SC con diagnóstico clínico. El caso aquí publicado sería el primero con diagnóstico molecular en Colombia y Latinoamérica.
El SC es una enfermedad de origen genético dada por daños en el gen VPS13B, antes denominado COH1, en el locus 8q22-23, que producen cambios en la proteína de su mismo nombre; por homología con modelos en levaduras se le atribuyen funciones relacionadas con el transporte intra e intercelular de proteínas mediado por vesículas. Estudios funcionales mostraron que la proteína VPS13B se localiza en el aparato de Golgi con la proteína de matriz GM130 y es necesaria para el mantenimiento, el ensamble de las cisternas y la regulación de la formación de túbulos de membrana. La ausencia o alteración de esta proteína puede llevar a la disrupción de la integridad del aparato de Golgi y de su funcionalidad; de igual manera, se han asociado las fallas en esta proteína a defectos de glicosilación y a daños en el mantenimiento de la vía lisosomal-endosomal alterando procesos de degradación intracelular. No está claro cómo se relacionan los defectos en la proteína VPS13B con los rasgos fenotípicos. Las diferencias en los fenotipos de los pacientes pueden atribuirse a distintas mutaciones en el gen o a las diferencias genéticas entre las poblaciones.
Mochida et al., describieron en 2004 una familia francesa con una mutación que cambiaba el marco de lectura y solo truncaba la región C terminal de la proteína dando lugar a una pérdida parcial en la función de VPS13B por una disrupción de la localización subcelular; este hallazgo se hizo en afectados por SC sin microcefalia. Además, se han descrito mutaciones específicas del gen relacionadas con trastornos del espectro autista y con discapacidad intelectual.
Dado que en el SC se han documentado diferentes etiologías genéticas como mutaciones puntuales, microdeleciones y microduplicaciones en homocigosis, heterocigosis y en combinación en el gen VPS13B, para el diagnóstico en los pacientes que cumplen con los criterios clínicos de SC se han propuesto diferentes técnicas moleculares.
El Chehadeh et al., en 2011 analizaron las publicaciones en que se describen diferentes técnicas moleculares para el diagnóstico de SC como MLPA (por la sigla en inglés de Multiplex ligation-dependent probe amplification), secuenciación del gen e hibridación genómica comparativa por microarreglos de alta resolución (HGCa); concluyeron que al menos en un tercio de las familias en las que se encuentra una causa genética del SC se observan variaciones en el número de copias de oligonucleótidos que incluyen el gen VPS13B y que se pueden detectar mediante HGCa de alta resolución (244K) y que no necesariamente se deben tener sondas específicas. Además, afirmaron que el MLPA con sondas específicas para VPS13B es eficiente para el diagnóstico de SC, pero solamente se hace en muy pocos laboratorios por la baja prevalencia del SC. Propusieron que la primera prueba que se haga en pacientes con sospecha clínica de SC sea la HCGa de alta resolución por la facilidad del acceso a ella en todo el mundo. El diagnóstico del paciente aquí reportado se hizo con dicha prueba y se encontró una deleción de 0.153 Mb que incluía el gen VP13B1.
Diagnóstico prenatal y consejo genético.- Es posible si se han identificado las mutaciones en los miembros de la familia. El SC se transmite siguiendo un patrón autosómico recesivo y se recomienda el consejo genético para individuos en riesgo.
Tratamiento.– Son necesario lentes para corregir la refracción y el estrabismo. La disfunción cognitiva precisa de una educación especial y los niños suelen asistir a escuelas especializadas. La logopedia es importante durante los años de educación preescolar para facilitar el desarrollo del habla, así como la fisioterapia para el retraso motor, la hipotonía y la torpeza motora. Las infecciones respiratorias deben tratarse con antibióticos. A algunos pacientes se les ha dado factor estimulante de colonias de granulocitos y monocitos o GM-CSF para tratar la neutropenia. Es necesaria una evaluación oftalmológica para determinar la agudeza visual. No se ha desarrollado ningún tratamiento efectivo para detener la progresión de la enfermedad de la retina.
Más tarde, puede ofrecerse formación específica para discapacitados visuales. Debe darse apoyo psicosocial a los pacientes y a sus familiares. La esperanza de vida no disminuye pero la calidad de vida se ve mermada por la discapacidad visual.
Bibliografía
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=193
- Cohen MM, Hall BD, Smith DW, Graham CB, Lampert KJ. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular and limb anomalies. J Pediatr. 1973; 83 (2): 280-4.
- Carey JC, Hall BD. Confirmation of the Cohen syndrome. J Pediatr. 1978 ; 93 (2): 239-44.
- Shearman JR, Wilton AN. A canine model of Cohen syndrome: Trapped Neutrophil Syndrome. BMC Genomics. 2011 ; 12: 258.
- Douzgou S, Petersen MB. Clínicasl variability of genetic isolates of Cohen syndrome. Clin Genet. 2011 ; 79 (6): 501-6. Falk MJ, Wang H, Traboulsi EI. Cohen Syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; c1993-2014 [ cited 23 Mar 2012 ] Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301655.
- Taban M, Memoracion-Peralta DS, Wang H, Al-Gazali LI, Traboulsi EI. Cohen syndrome: report of nine cases and review of the literature, with emphasis on ophthalmic features. J AAPOS. 2007 ; 11 (5): 431-7.
- Pirgon O, Atabek ME, Sert A. Metabolic syndrome manifestations in Cohen syndrome: description of two new patients. J Child Neurol. 2006 ; 21 (6): 536-8.
- Falk MJ, Feiler HS, Neilson DE, Maxwell K, Lee JV, Segall SK, Robin NH, et al. Cohen syndrome in the Ohio Amish. Am J Med Genet A. 2004 ; 128A (1): 23-8.
- El Chehadeh-Djebbar S, Faivre L, Moncla A, Aral B, Missirian C, Popovici C, et al. The power of high-resolution non-targeted array-CGH in identifying intragenic rearrangements responsible for Cohen syndrome. J Med Genet. 2011 Nov;48(11):e1. DOI 10.1136/jmg.2011.088948.
- Mochida GH, Rajab A, Eyaid W, Lu A, Al-Nouri D, Kosaki K, et al. Broader geographical spectrum of Cohen syndrome due to COH1 mutations. J Med Genet. 2004 Jun;41(6):e87.
- Chandler KE, Kidd A, Al-Gazali L, Kolehmainen J, Lehesjoki AE, Black GC, et al. Diagnostic criteria, clínicasl characteristics, and natural history of Cohen syndrome. J Med Genet. 2003 Apr;40(4):233-41.
- Kolehmainen J, Wilkinson R, Lehesjoki AE, Chandler K, Kivitie-Kallio S, Clayton-Smith J, et al. Delineation of Cohen syndrome following a large-scale genotype-phenotype screen. Am J Hum Genet. 2004 Jul;75(1):122-7.
- Aitken KJ. Cohen syndrome. In: An A-Z of genetic factors in autism : a handbook for professionals. London: Jessica Kingsley; 2010. p. 164-5.
- Rim PH, Figueirêdo ES, Hirata FE, Steiner CE, Marques-de-Faria AP. Ocular findings in Brazilian identical twins with Cohen syndrome: case report. Arq Bras Oftalmol. 2009 Nov-Dec;72(6):815-8.
- Santana Hernández EE, Tamayo Chang VJ. Síndrome de Cohen. Presentación de un caso. Medisur. 2014 Abr;12(2):437-41.
- Wang H, Falk MJ, Wensel C, Traboulsi EI. Cohen Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from http:// www.ncbi.nlm.nih.gov/books/NBK1482/
- Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Träskelin AL, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003 Jun;72(6):1359-69.
- Falk MJ, Feiler HS, Neilson DE, Maxwell K, Lee JV, Segall SK, et al. Cohen syndrome in the Ohio Amish. Am J Med Genet A. 2004 Jul;128A(1):23-8.
- Rafiq MA, Leblond CS, Saqib MA, Vincent AK, Ambalavanan A, Khan FS, et al. Novel VPS13B Mutations in Three Large Pakistani Cohen Syndrome Families Suggests a Baloch Variant with Autistic-Like Features. BMC Med Genet. 2015 Jun 25;16:41. DOI 10.1186/s12881-015-0183-0.
- Horn D, Krebsová A, Kunze J, Reis A. Homozygosity mapping in a family with microcephaly, mental retardation, and short stature to a Cohen syndrome region on 8q21.3-8q22.1: redefining a clínicasl entity. Am J Med Genet. 2000 Jun;92(4):285-92.
- Rivera-Brugués N, Albrecht B, Wieczorek D, Schmidt H, Keller T, Göhring I, et al. Cohen syndrome diagnosis using whole genome arrays. J Med Genet. 2011 Feb;48(2):136-40. DOI 10.1136/jmg.2010.082206.